
The following pages include guidance for management of patients with HCV in unique and key populations.
This section provides guidance on the treatment of chronic HCV infection in HIV/HCV-coinfected patients. For guidance regarding HIV/HCV-coinfected individuals with acute HCV infection, please see the Acute HCV section. HIV/HCV-coinfected patients suffer from more liver-related morbidity and mortality, nonhepatic organ dysfunction, and overall mortality than HCV-monoinfected patients (Lo Re, 2014); (Chen, 2009). Even in the potent HIV antiretroviral therapy (ART) era, HIV infection remains independently associated with advanced liver fibrosis and cirrhosis in patients with HIV/HCV coinfection (Fierer, 2013); (Kirk, 2013); (de Ledinghen, 2008); (Thein, 2008a). As such, HCV treatment in HIV-infected patients should be a priority for providers, payers, and patients. If HCV treatment is delayed for any reason, however, liver disease progression should be monitored at routine intervals as recommended in the guidance (see When and in Whom to Initiate Therapy, recommendation for repeat liver disease assessment).
With the availability of HCV direct-acting antivirals (DAAs), efficacy and adverse event rates among persons with HIV/HCV coinfection are similar to those observed with HCV monoinfection (Rockstroh, 2018); (Bhattacharya, 2017); (Wyles, 2017b); (Naggie, 2015); (Rockstroh, 2015); (Sulkowski, 2015); (Wyles, 2015). Simplified HCV treatment has also been demonstrated to be effective in persons living with HIV. Recent data from a global sample of patients undergoing antiviral treatment for chronic HCV infection (MINMON study) suggested that a minimal monitoring approach was safe and achieved SVR at a rate comparable to that with standard monitoring (see Monitoring Patients Who Are Starting HCV Treatment, Are on Treatment, or Have Completed Therapy ). Of the 400 participants, 399 initiated sofosbuvir/velpatasvir treatment. At entry, 166 (42%) were living with HIV, and 94.6% achieved SVR, similar to those without HIV (95.3%) (Solomon, 2022). In addition to other exclusion criteria to simplified treatment, individuals receiving a TDF-containing regimen with eGFR < 60 ml/min should not receive simplified HCV treatment given the need for additional monitoring.
Treatment of HIV/HCV-coinfected patients, however, requires continued awareness and attention to the complex drug-drug interactions that can occur between DAAs and antiretroviral medications. Drug interactions with DAAs and antiretroviral agents are summarized in the text and tables of this section as well as in the US Department of Health and Human Services HIV treatment guidelines (https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv/whats-new-guidelines). The University of Liverpool drug interactions website (www.hep-druginteractions.org) is another resource for screening for drug-drug interactions with DAAs. Drug interactions should be carefully reviewed before proceeding with simplified HCV treatment in HIV/HCV coinfection.
Due to shared modes of transmission, HIV/HCV-coinfected patients are at risk for hepatitis B virus (HBV) infection. HBV reactivation has been reported in patients starting DAA HCV therapy who are not on active HBV agents. Consistent with general recommendations for the assessment of both HIV- and HCV-infected patients, all patients initiating HCV DAA therapy should be assessed for HBV coinfection with HBsAg, anti-HBs, and anti-HBc testing. HIV-infected patients with evidence of HBV infection should be on antiretroviral agents with activity against HBV, preferably tenofovir disoproxil fumarate or tenofovir alafenamide. For patients who are only anti-HBc positive and not on tenofovir-based ART, subsequent monitoring for HBV reactivation should be conducted as detailed in the Monitoring section.
Recommendations Related to HCV Medication Interactions With HIV Antiretroviral Medications |
|
|---|---|
| RECOMMENDED |
RATING |
| Antiretroviral drug switches, when needed, should be done in collaboration with the HIV practitioner. For HIV antiretroviral and HCV direct-acting antiviral combinations not addressed below, expert consultation is recommended. | I, A |
|
Daily fixed-dose combination of elbasvir (50 mg)/grazoprevir (100 mg) Elbasvir/grazoprevir should be used with antiretroviral drugs with which it does not have clinically significant interactions: abacavir, bictegravir, cabotegravir, dolutegravir, doravirine, emtricitabine, ibalizumab-uiyk, lamivudine, maraviroc, raltegravir, rilpivirine, and tenofovir. |
IIa, B |
|
Daily fixed-dose combination of glecaprevir (300 mg)/pibrentasvir (120 mg)a Glecaprevir/pibrentasvir should be used with antiretroviral drugs with which it does not have clinically significant interactions: abacavir, bictegravir, cabotegravir, dolutegravir, doravirine, emtricitabine, fostemsavir, ibalizumab-uiyk, lamivudine, maraviroc, raltegravir, rilpivirine, and tenofovir. Given the increase in glecaprevir exposures and limited data on the safety of elvitegravir/cobicistat with glecaprevir/pibrentasvir, monitoring for hepatic toxicity is recommended until additional safety data are available in HIV/HCV-coinfected patients. |
IIa, B |
|
Daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg) Sofosbuvir/velpatasvir can be used with most antiretrovirals but not efavirenz, etravirine, or nevirapine. Because tenofovir levels, when given as tenofovir disoproxil fumarate, may increase with sofosbuvir/velpatasvir, concomitant use mandates consideration of renal function and should be avoided in those with an eGFR <60 mL/min. Due to limited experience with this drug combination, renal monitoring is recommended in patients taking tenofovir disoproxil fumarate and cobicistat or ritonavir with sofosbuvir/velpatasvir. Tenofovir alafenamide may be an alternative to tenofovir disoproxil fumarate during sofosbuvir/velpatasvir treatment for patients who take cobicistat or ritonavir as part of their antiretroviral therapy. |
IIa, B |
|
Daily fixed-dose combination of ledipasvir (90 mg)/sofosbuvir (400 mg) Ledipasvir/sofosbuvir can be used with most antiretrovirals. Because this therapy increases tenofovir levels when given as tenofovir disoproxil fumarate, concomitant use mandates consideration of renal function and should be avoided in those with an eGFR <60 mL/min. Absolute tenofovir levels are highest and may exceed exposures for which there are established renal safety data when tenofovir disoproxil fumarate is administered with ritonavir- or cobicistat-containing regimens. Due to lack of sufficient safety data with this drug combination, consideration should be given to changing the antiretroviral regimen. If the combination is used, renal monitoring is recommended during the dosing period. Tenofovir alafenamide may be an alternative to tenofovir disoproxil fumarate during ledipasvir/sofosbuvir treatment for patients taking cobicistat or ritonavir as part of their antiretroviral therapy. |
IIa, C |
| For combinations including tenofovir disoproxil fumarate wherein increased tenofovir levels are expected, baseline and ongoing assessment for tenofovir nephrotoxicity is recommended. | IIa, C |
|
Daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg)/voxilaprevir (100 mg) Sofosbuvir/velpatasvir/voxilaprevir should be used with antiretroviral drugs with which they do not have substantial interactions: abacavir, bictegravir, cabotegravir, dolutegravir, doravirine, emtricitabine, ibalizumab-uiyk, lamivudine, maraviroc, raltegravir, rilpivirine, and tenofovir alafenamide. Given increases in voxilaprevir AUC with darunavir/ritonavir or elvitegravir/cobicistat coadministration and lack of clinical safety data, monitoring for hepatic toxicity is recommended until additional safety data are available in HIV/HCV-coinfected patients. Because this therapy has the potential to increase tenofovir levels when given as tenofovir disoproxil fumarate, concomitant use mandates consideration of renal function and should be avoided in those with an eGFR <60 mL/min. In patients concomitantly receiving sofosbuvir/velpatasvir/voxilaprevir and tenofovir disoproxil fumarate, renal monitoring is recommended during the dosing period. |
IIa, B |
| a Dosing is 3 coformulated tablets (glecaprevir [100 mg]/pibrentasvir [40 mg]) taken once daily. Please refer to the prescribing information. | |
Extensive recommendations for ART use (including for persons anticipating HCV treatment) are available at https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv/whats-new-guidelines.
Antiretroviral drug switches may be performed to allow compatibility with DAAs with the goal of maintaining HIV suppression without compromising future options. Considerations include prior treatment history, response(s) to ART, resistance profiles, and drug tolerance (DHHS, 2021); (Gunthard, 2014). Treatment interruption in HIV/HCV-coinfected individuals is not recommended as it is associated with increased cardiovascular events (SMART, 2006) and increased rates of fibrosis progression and liver-related events (Thorpe, 2011); (Tedaldi, 2008). The availability of multiple effective HCV DAA and HIV antiretroviral regimens makes it possible for all HIV/HCV-coinfected patients to safely and successfully receive HCV treatment. Switching an optimized antiretroviral regimen carries risks, including adverse effects and HIV viral breakthrough (Eron, 2010). HIV viral breakthrough is of particular concern for those with substantial antiretroviral experience or known resistance to antiretroviral drugs. If necessary, ART changes should be undertaken in close collaboration with the treating HIV provider prior to HCV treatment initiation.
Although fewer HIV/HCV-coinfected patients than HCV-monoinfected patients have been treated in DAA trials, efficacy rates to date have been remarkably similar between the groups (Rockstroh, 2018); (Dieterich, 2015); (Naggie, 2015); (Osinusi, 2015); (Rockstroh, 2015); (Rodriguez-Torres, 2015); (Sulkowski, 2015); (Wyles, 2015); (Wyles, 2015b); (Dieterich, 2014b); (Sulkowski, 2014); (Sulkowski, 2013). Thus, results from HCV monoinfection studies largely justify the recommendations for HIV/HCV coinfection (discussed in the Initial Treatment, and Retreatment sections), which are generally similar to HCV monoinfection. Discussion specific to HIV/HCV coinfection research is included here.
In general, few HIV/HCV-coinfected patients with compensated cirrhosis have been included in clinical trials of DAAs, and no data are available regarding HIV/HCV-coinfected patients with renal insufficiency or who have undergone solid organ transplantation. Despite the lack of data, it is highly likely that response rates are similar to those of HCV-monoinfected patients because no study to date in the DAA era has shown a lower efficacy for HIV/HCV-coinfected patients. Therefore, the respective guidance from these sections should be followed if treatment is otherwise warranted, with consideration of drug-drug interactions.
The safety, tolerability, and efficacy of the second-generation NS3/4A serine protease inhibitor grazoprevir plus the NS5A inhibitor elbasvir were assessed in patients with HIV/HCV coinfection in the C-EDGE COINFECTION study. C-EDGE COINFECTION was a phase 3, nonrandomized, open-label, single-arm study in which treatment-naive patients with genotype 1, 4, or 6 and HIV coinfection (with or without compensated cirrhosis) were enrolled in Europe, the US, and Australia (Rockstroh, 2015). All patients were either naive to treatment with any ART and a CD4 cell count >500/mm3 (n=7), or stable on current ART for at least 8 weeks with a CD4 cell count >200/mm3 (n=211) and undetectable HIV RNA. All 218 enrolled patients received the once-daily fixed-dose combination of elbasvir (50 mg) plus grazoprevir (100 mg) for 12 weeks. All 218 patients completed follow-up at week 12. The median baseline CD4 cell count was 568/mm3 (range, 424-626/mm3). Limited antiretroviral regimens were allowed, specifically a nucleoside/nucleotide backbone of abacavir (21.6%) or tenofovir (75.2%) in combination with raltegravir (52%), dolutegravir (27%), or rilpivirine (17%).
SVR12 was achieved by 96% (210/218) of patients (95% CI, 92.9-98.4). One patient did not achieve SVR12 for a nonvirologic reason and 7 patients without cirrhosis relapsed (2 subsequently confirmed as reinfections, highlighting the requirement of continued harm-reduction strategies after SVR). Thirty-five patients with compensated cirrhosis achieved SVR12. The most common adverse events were fatigue (13%; n=29), headache (12%; n=27), and nausea (9%; n=20). No patient discontinued treatment because of an adverse event. Three out of 6 patients who relapsed before SVR12 had NS3 and/or NS5A resistance-associated substitutions (RASs) while the others had wild type virus at the time of relapse. Two patients receiving ART had transient HIV viremia but subsequently returned to undetectable levels without a change in ART. No significant changes were observed with CD4 cell counts or new opportunistic infections. Elbasvir/grazoprevir without ribavirin appears effective and well tolerated among patients coinfected with HIV, with or without compensated cirrhosis. These data are consistent with previous trials of this regimen in the HCV monoinfected population (Zeuzem, 2017).
Pharmacology and Drug Interaction Data
Elbasvir is a substrate for CYP3A4 and the efflux transporter P-glycoprotein (P-gp). Grazoprevir is a substrate for CYP3A4, P-gp, and the liver uptake transporter OATP1B1. Moderate and strong CYP3A and P-gp inducers (including efavirenz) are not recommended for coadministration with elbasvir/grazoprevir. OATP1B1 inhibitors are also not recommended with grazoprevir.
Elbasvir/grazoprevir is not compatible with any ritonavir- or cobicistat-boosted HIV protease inhibitor, elvitegravir/cobicistat, efavirenz, etravirine, or nevirapine (Feng, 2019). Drug interaction studies showed no clinically significant interactions between elbasvir/grazoprevir and dolutegravir, raltegravir, doravirine, rilpivirine, or tenofovir disoproxil fumarate (Ankrom, 2019); (Feng, 2019a); (Feng, 2019b); (Yeh, 2015b).
The safety and efficacy of glecaprevir (a pangenotypic NS3/4A protease inhibitor) coformulated with pibrentasvir (a pangenotypic NS5A inhibitor) were evaluated in the phase 3, multicenter EXPEDITION-2 study (Rockstroh, 2018). This study evaluated 8 weeks of the daily fixed-dose combination of glecaprevir (300 mg)/pibrentasvir (120 mg) among 137 HIV/HCV-coinfected adults without cirrhosis, and 12 weeks of glecaprevir/pibrentasvir in 16 HIV/HCV-coinfected patients with compensated cirrhosis. Treatment-naive and -experienced patients with genotype 1, 2, 3, 4, or 6 infection were enrolled. Patients were either antiretroviral naive with a CD4 cell count ≥500/mm3, or on a stable ART regimen for at least 8 weeks with a CD4 cell count ≥200/mm3. ART drugs included raltegravir, dolutegravir, rilpivirine, tenofovir disoproxil fumarate, tenofovir alafenamide, abacavir, emtricitabine, and lamivudine. One patient received elvitegravir/cobicistat. Overall SVR12 was 98% (136/136 among those without cirrhosis on the 8-week regimen, and 14/15 in those with compensated cirrhosis on the 12-week regimen). Four serious adverse events were reported, none of which were DAA related. One of these led to treatment discontinuation.
The EXPEDITION-8 trial found that 8 weeks of glecaprevir/pibrentasvir achieved similar SVR rates to those achieved with 12 weeks of treatment in treatment-naive patients with cirrhosis (Brown, 2020). While persons with HIV were not included in this study, SVR rates are likely to be similar in PWH.
Pharmacology and Drug Interaction Data
Glecaprevir is metabolized by CYP3A as a secondary pathway, and glecaprevir and pibrentasvir are substrates for P-gp and breast cancer resistance protein (BCRP). Glecaprevir is also a substrate for the hepatic uptake transporter organic anion-transporting polypeptide (OATP) 1B1/3. Glecaprevir and pibrentasvir are weak inhibitors of CYP3A, CYP1A2, and uridine glucuronosyltransferase (UGT) 1A1. Glecaprevir and pibrentasvir inhibit P-gp, BCRP, and OATP1B1/3. Compounds that inhibit P-gp, BCRP, or OATP1B1/3 may increase glecaprevir and pibrentasvir concentrations. In contrast, drugs that induce P-gp/CYP3A may decrease glecaprevir and pibrentasvir concentrations.
Glecaprevir and pibrentasvir area under the curve (AUC) are increased roughly 3-fold and 1.57-fold, respectively, with tenofovir alafenamide/emtricitabine/elvitegravir/cobicistat (Kosloski, 2020). A single patient received this combination in the EXPEDITION-2 study. Although the increases in AUC of glecaprevir and pibrentasvir when coadministered with elvitegravir/cobicistat are not considered clinically relevant by the manufacturer or the US Food and Drug Administration (FDA), due to lack of sufficient clinical safety data, close monitoring for hepatic toxicity is recommended until additional safety data are available in HIV/HCV-coinfected patients. Consider liver enzyme testing every 4 weeks.
No clinically significant interactions were observed with glecaprevir/pibrentasvir in a drug interaction study with dolutegravir, raltegravir, rilpirivine, abacavir, lamivudine, emtricitabine, or tenofovir (Kosloski, 2020). Boosted protease inhibitors are not recommended with glecaprevir/pibrentasvir. Glecaprevir and pibrentasvir exposures were both at least 47% lower when coadministered with efavirenz compared to observed concentrations when given alone in other studies and, therefore, concomitant use is not recommended (Kosloski, 2020). Etravirine and nevirapine should not be used due to the potential for decreased glecaprevir/pibrentasvir exposures.
Glecaprevir absorption is pH dependent and glecaprevir exposures are reduced approximately 50% with 40 mg of omeprazole daily. Despite the reduced glecaprevir exposures, pooled data from the phase 2/3 glecaprevir/pibrentasvir trials found that patients receiving proton pump inhibitors had similar SVR rates compared to patients not receiving a gastric acid modifier (Flamm, 2019).
The safety and efficacy of 12 weeks of ledipasvir/sofosbuvir were evaluated in the phase 2, single-center, open-label ERADICATE trial, which included 50 HIV/HCV-coinfected patients with genotype 1 infection who were treatment naive without cirrhosis (Osinusi, 2015). Thirteen patients were not receiving ART and 37 patients were on protocol-allowed medications (tenofovir, emtricitabine, rilpivirine, raltegravir, and efavirenz). Although the inclusion criteria for patients receiving ART allowed CD4 cell counts >100/mm3, the median CD4 cell count was 576/mm3. Overall, 98% achieved SVR12 (13/13 in the treatment-naive arm and 36/37 in the treatment-experienced arm). There were no deaths, discontinuations, or clinically significant, serious adverse events. Renal function was monitored frequently during this trial and after administration of study drugs using a battery of tests (serum creatinine, eGFR, urinary beta-2 microglobulin, and urine protein and glucose). No clinically significant changes in these parameters or renal toxicity were observed.
A larger study, ION-4, reported similar outcomes with ledipasvir/sofosbuvir (Naggie, 2015). A total of 335 HCV treatment-naive and -experienced HIV/HCV-coinfected patients were enrolled in the study and received ledipasvir/sofosbuvir once daily for 12 weeks. Patients received tenofovir disoproxil fumarate and emtricitabine with raltegravir (44%), efavirenz (48%), or rilpivirine (9%). Genotypes included were 1a (75%), 1b (23%), and 4 (2%). Twenty percent of patients had compensated cirrhosis, 34% were black, and 55% had not responded to prior HCV treatment. The overall SVR12 was 96% (321/335). Two patients had on-treatment virologic failure judged to be the result of nonadherence; 10 had virologic relapse after discontinuing treatment; 1 died from endocarditis associated with injection drug use; and 1 was lost to follow-up. SVR12 rates were 94% (63/67) among patients with compensated cirrhosis and 97% (179/185) among treatment-experienced patients. No patients discontinued the study drugs because of an adverse event. Although all patients had an eGFR >60 mL/min at study entry, drug interaction studies suggested that patients receiving tenofovir disoproxil fumarate could have increased tenofovir levels. There were 4 patients in whom serum creatinine level rose to ≥0.4 mg/dL. Two remained on tenofovir disoproxil fumarate, one had the tenofovir disoproxil fumarate dose reduced, and the other stopped taking tenofovir disoproxil fumarate.
Neither the ERADICATE nor the ION-4 study investigators reported clinically significant changes in CD4 cell counts or HIV RNA levels. Thus, these data suggest that 12 weeks of ledipasvir/sofosbuvir is a safe and effective regimen for HIV/HCV-coinfected patients with genotype 1 infection taking selected ART (Naggie, 2015); (Osinusi, 2015). There are limited data regarding an 8-week course of ledipasvir/sofosbuvir in HIV/HCV-coinfected patients (Vega, 2019); (Isakov, 2018); (Ingiliz, 2016). Additionally, clinical trial data of daclatasvir (an NS5A inhibitor similar to ledipasvir) plus sofosbuvir in HIV/HCV-coinfected patients demonstrated a lower SVR rate (76%) with 8 weeks of treatment compared to 12 weeks (97%) (Wyles, 2015). Therefore, a shortened treatment course for HIV/HCV-coinfected persons is not recommended at this time.
Pharmacology and Drug Interaction Data
Ledipasvir and sofosbuvir are P-gp and BCRP substrates; ledipasvir is also an inhibitor of both P-gp and BCRP transporters. Ledipasvir absorption is pH dependent. Refer to product labeling for guidance on temporal separation and dosing of gastric acid modifying agents.
Drug interaction studies of ledipasvir (with or without sofosbuvir) with antiretroviral drugs in uninfected persons did not identify clinically significant interactions with abacavir, bictegravir, dolutegravir, doravirine, emtricitabine, lamivudine, raltegravir, rilpivirine, or tenofovir alafanamide (Ankrom, 2019); (Garrison, 2018); (Garrison, 2015); (German, 2014). Ledipasvir AUC is decreased by 34% when coadministered with efavirenz-containing regimens and increased by 96% when coadministered with ritonavir-boosted atazanavir (German, 2014). No dose adjustments of ledipasvir are recommended to account for these interactions.
Ledipasvir/sofosbuvir increases tenofovir levels when given as tenofovir disoproxil fumarate, which may increase the risk of tenofovir-associated renal toxicity. This combination should be avoided in patients with an eGFR <60 mL/min. With the addition of ledipasvir/sofosbuvir, tenofovir levels (when given as tenofovir disoproxil fumarate) are increased with efavirenz, rilpivirine (German, 2014), dolutegravir, ritonavir-boosted atazanavir, and ritonavir-boosted darunavir (German, 2015). The absolute tenofovir levels are highest and may exceed exposures for which there are established renal safety data when tenofovir disoproxil fumarate is administered with ritonavir- or cobicistat-containing regimens; consideration should be given to changing the antiretroviral regimen. Tenofovir alafenamide may be an alternative to tenofovir disoproxil fumarate during ledipasvir/sofosbuvir treatment for patients who take cobicistat or ritonavir as part of their ART.
In patients with an eGFR <60 mL/min who are taking tenofovir disoproxil fumarate with ledipasvir/sofosbuvir, renal parameters should be checked at baseline and monthly while on ledipasvir/sofosbuvir. Baseline parameters should include creatinine level, electrolytes (including phosphorus), and urinary protein and glucose according to recent guidelines for the management of chronic kidney disease in those with HIV, which include indications for nephrology consultation (Lucas, 2014). A change in ART should be considered for those at high risk for renal toxicity—especially those with an eGFR between 30 mL/min and 60 mL/min or who have preexisting evidence of Fanconi syndrome, and particularly those taking tenofovir disoproxil fumarate and a ritonavir- or cobicistat-containing regimen. Tenofovir disoproxil fumarate should also be properly dosed and adjusted for eGFR at baseline and while on therapy (Lucas, 2014).
Data are limited regarding the renal safety of tenofovir when given as tenofovir alafenamide with ledipasvir/sofosbuvir. However, a small pharmacokinetic study among persons with HIV on a boosted protease inhibitor and tenofovir alafenamide containing regimen found that the addition of ledipasvir/sofosbuvir did not worsen renal biomarkers (Brooks, 2020b). A study of tenofovir pharmacokinetics in healthy volunteers receiving the combination of tenofovir alafenamide, emtricitabine, and cobicistat-boosted elvitegravir with ledipasvir/sofosbuvir found that tenofovir levels were only 20% of the typical tenofovir exposures seen with tenofovir disoproxil fumarate (Garrison, 2015). Based on these pharmacokinetic data in healthy volunteers, tenofovir alafenamide may be an alternative to tenofovir disoproxil fumarate during ledipasvir/sofosbuvir treatment for patients on ritonavir- or cobicistat-containing regimens.
The safety and efficacy of 12 weeks of sofosbuvir/velpatasvir were evaluated in a phase 3 study among 106 ART-controlled, HIV/HCV-coinfected patients (Wyles, 2017b). Patients with genotype 1, 2, 3, or 4 infection were included; 18% (19/106) had compensated cirrhosis. HIV was controlled on ART including non-nucleoside reverse-transcriptase inhibitor- (rilpivirine), integrase inhibitor- (raltegravir or elvitegravir/cobicistat), or ritonavir-boosted protease inhibitor- (atazanavir, lopinavir, or darunavir) based regimens with either tenofovir/emtricitabine or abacavir/lamivudine. Fifty-three percent (56/106) of participants were on tenofovir disoproxil fumarate with a pharmacologic boosting agent (ritonavir or cobicistat). Neither efavirenz nor etravirine were allowed in this study as concomitant dosing with sofosbuvir/velpatasvir in healthy volunteers resulted in clinically significant decreases in velpatasvir exposure. SVR12 was 95% with 2 relapses, both occurring in genotype 1a-infected patients. Similar results were noted in patients with compensated cirrhosis and in those with baseline NS5A RASs (n=12 at 15% threshold; SVR12=100%). There were no clinically significant changes in serum creatinine or eGFR, and no patients required a change in their ART during the study period.
Pharmacology and Drug Interaction Data
Velpatasvir is available only in a fixed-dose combination tablet with sofosbuvir. Velpatasvir is metabolized by CYP3A4, CYP2C8, and CYP2B6. It does not appear to inhibit or induce any CYP enzymes. Velpatasvir is a substrate for P-gp and BCRP, and inhibits P-gp, BCRP, and OATP1B1/1B3/2B1 but does not induce any transporters.
Velpatasvir absorption is pH dependent. Refer to product labeling for guidance on temporal separation and dosing of gastric acid modifying agents.
Drug interaction studies with sofosbuvir/velpatasvir have been performed in HIV and HCV seronegative volunteers. As with ledipasvir/sofosbuvir, tenofovir exposures are increased, which may be problematic for individuals with an eGFR <60 mL/min or in those receiving ritonavir- or cobicistat-containing ART with tenofovir disoproxil fumarate. Fifty-six HIV/HCV-coinfected individuals receiving the combination of tenofovir disoproxil fumarate with ritonavir- or cobicistat-containing ART were treated with sofosbuvir/velpatasvir in the ASTRAL-5 study with no difference in median creatinine clearance before and after sofosbuvir/velpatasvir treatment, however, poor renal function was an exclusion for this study (Wyles, 2017b). In individuals with an eGFR <60 mL/min and those requiring ritonavir- or cobicistat-containing ART, consider use of tenofovir alafenamide in place of tenofovir disoproxil fumarate. If the combination of tenofovir disoproxil fumarate with a ritonavir- or cobicistat-containing ART is required or in those with an eGFR <60 mL/min, renal parameters should be checked at baseline and monthly while on sofosbuvir/velpatasvir.
Based on data from healthy volunteers, tenofovir pharmacokinetics are lower with tenofovir alafenamide relative to tenofovir disoproxil fumarate. Thus, tenofovir alafenamide may be an alternative to tenofovir disoproxil fumarate during sofosbuvir/velpatasvir treatment for patients who take cobicistat or ritonavir as part of their ART. However, there are no safety data for this combination in HIV/HCV-coinfected patients.
Drug-drug interaction studies in healthy volunteers found no clinically significant interaction between sofosbuvir/velpatasvir and atazanavir/ritonavir, bictegravir, darunavir/ritonavir, dolutegravir, elvitegravir/cobicistat, lopinavir/ritonavir, raltegravir, rilpivirine, emtricitabine, or tenofovir alafenamide (Garrison, 2018); (Mogalian, 2018). Velpatasvir exposures are significantly reduced with efavirenz and this combination is not recommended. Etravirine and nevirapine have not been studied with sofosbuvir/velpatasvir but are also not recommended. Indirect bilirubin level increases have been reported when sofosbuvir/velpatasvir was used in patients on atazanavir/ritonavir. These changes are not considered clinically significant.
The data supporting use of sofosbuvir/velpatasvir/voxilaprevir are described in the Initial Treatment of HCV Infection and Retreatment of Persons in Whom Prior Therapy Has Failed sections. There are limited data on sofosbuvir/velpatasvir/voxilaprevir in HIV/HCV-coinfected patients. The RESOLVE study included 17 individuals with HIV coinfection and a previous DAA treatment failure (Wilson, 2019). SVR12 was 82% by intention-to-treat analysis and 93% by per protocol analysis. While these data are limited, they suggest response rates in HIV/HCV-coinfected patients are similar to those of HCV-monoinfected patients. Therefore, the respective guidance from the aforementioned treatment and retreatment sections should be followed, with consideration of drug-drug interactions.
Pharmacology and Drug Interaction Data
Voxilaprevir is a substrate for P-gp, OATP1B1/3, BCRP, CYP3A, CYP1A2, and CYP2C8. Voxilaprevir inhibits OATP1B1/3, P-gp, and BCRP. Voxilaprevir AUC is increased 331% with ritonavir-boosted atazanavir and this combination is not recommended (Garrison, 2017). Voxilaprevir AUC is increased 171% with tenofovir alafenamide/emtricitabine/elvitegravir/cobicistat, and 143% with tenofovir disoproxil fumarate/emtricitabine and ritonavir-boosted darunavir. Although these increases in voxilaprevir AUC were not deemed clinically relevant by the manufacturer or the FDA, due to lack of clinical safety data, close monitoring for hepatic toxicity is recommended until additional safety data are available in HIV/HCV-coinfected patients. Consider liver enzyme testing every 4 weeks.
Velpatasvir absorption is pH dependent. Velpatasvir AUC is reduced approximately 50% when given with omeprazole 20 mg daily as part of the fixed-dose sofosbuvir/velpatasvir/voxilaprevir combination. Refer to product labeling for guidance on temporal separation and dosing of gastric acid modifying agents.
Tenofovir concentrations are increased with sofosbuvir/velpatasvir/voxilaprevir when given as tenofovir disoproxil fumarate (Garrison, 2017). In individuals with an eGFR <60 mL/min, consider use of tenofovir alafenamide in place of tenofovir disoproxil fumarate in those requiring ritonavir- or cobicistat-containing ART. No substantial interactions were observed with bictegravir, emtricitabine, or rilpivirine.
Table. Drug Interactions Between Direct-Acting Antivirals and Antiretroviral Drugs—Recommended Regimens
Ledipasvir/ Sofosbuvir(LDV/SOF) |
Sofosbuvir/ Velpatasvir(SOF/VEL) |
Elbasvir/ Grazoprevir(ELB/GRZ) |
Glecaprevir/ Pibrentasvir(GLE/PIB) |
Sofosbuvir/ Velpatasvir/ Voxilaprevir(SOF/VEL/VOX) |
||
|---|---|---|---|---|---|---|
Protease
|
Boosted Atazanavir |
A | A | |||
Boosted Darunavir |
A | A | ||||
Boosted Lopinavir |
ND, A | A | ND | |||
NNRTIs |
Doravirine |
ND | ND | ND | ||
Efavirenz |
ND | ND | ||||
Rilpivirine |
||||||
Etravirine |
ND | ND | ND | ND | ND | |
Integrase
|
Bictegravir |
ND | ND | |||
Cabotegravir |
ND | ND | ND | ND | ND | |
Cobicistat-boosted elvitegravir |
C | C | C | |||
Dolutegravir |
ND | |||||
Raltegravir |
ND | |||||
Entry
|
Fostemsavir |
ND | ND | ND | ND | ND |
Ibalizumab-uiyk |
ND | ND | ND | ND | ND | |
Maraviroc |
ND | ND | ND | ND | ND | |
NRTIs |
Abacavir |
ND | ND | ND | ||
Emtricitabine |
||||||
Lamivudine |
ND | ND | ND | |||
Tenofovir disoproxil fumarate |
B, C | B, C | C | |||
Tenofovir alafenamide |
D | D | ND | D | ||
|
Green indicates coadministration is safe; yellow indicates a dose change or additional monitoring is warranted; and red indicates the combination should be avoided.
ND: No data For antiretroviral agents not included in the table above, please refer to the US Department of Health and Human Services HIV treatment guidelines (https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv/whats-new-guidelines) and/or the University of Liverpool drug interactions website (www.hep-druginteractions.org). |
||||||
Treatment Recommendations for Patients With HIV/HCV Coinfection |
|
|---|---|
| RECOMMENDED |
RATING |
|
HIV/HCV-coinfected persons should be treated and retreated the same as persons without HIV infection, after recognizing and managing interactions with antiretroviral medications (see Initial Treatment of HCV Infection and Retreatment of Persons in Whom Prior Therapy Has Failed). |
I, B |
Recommended for All Patients With HCV Infection Who Have Decompensated Cirrhosis
|
|
|---|---|
| RECOMMENDED |
RATING |
| Patients with HCV infection who have decompensated cirrhosis—moderate or severe hepatic impairment, ie, Child-Turcotte-Pugh (CTP) class B or class C—should be referred to a medical practitioner with expertise in that condition, ideally in a liver transplant center. | I, C |
Clinical trial data demonstrate that in the population of persons with decompensated cirrhosis, most patients receiving direct-acting antiviral (DAA) therapy experience improvement in clinical and biochemical indicators of liver disease between baseline and posttreatment week 12, including patients with CTP class C cirrhosis (Manns, 2016); (Welzel, 2016); (Charlton, 2015); (Curry, 2015). Improvements, however, may be insufficient to avoid liver-related death or the need for liver transplantation (Belli, 2016), highlighting that not everyone benefits from DAA therapy (Fernandez-Carrillo, 2016). Most deaths among those receiving DAA therapy relate to the severity of the underlying liver disease. Predictors of improvement or decline have not been clearly identified, although patients with a Model for End-Stage Liver Disease (MELD) score >20 or severe portal hypertension complications may be less likely to improve and might be better served by transplantation than antiviral treatment (El-Sherif, 2018); (Terrault, 2017); (Belli, 2016).
Real-world data comparing DAA response rates demonstrate that patients with cirrhosis and hepatocellular carcinoma (HCC) have lower SVR rates than cirrhotic patients without HCC (Beste, 2017); (Prenner, 2017). In a large VA study including sofosbuvir, ledipasvir/sofosbuvir, and paritaprevir/ritonavir/ombitasvir plus dasabuvir regimens (± ribavirin), overall SVR rates were 91% in patients without HCC versus 74% in those with HCC (Beste, 2017). After adjusting for confounders, the presence of HCC was associated with a lower likelihood of SVR (AOR=0.38). Whether this lower SVR can be overcome with an extended duration of therapy is unknown.
In a real-world study, DAA-induced SVR was associated with reduced risk of clinical disease progression in patients with Child-Pugh A cirrhosis but not in those with Child-Pugh B/C cirrhosis. A ≥2 point decrease in MELD score among patients with Child-Pugh B/C cirrhosis was not associated with improved clinical outcome (Krassenburg, 2021). In a large, multicenter, real-world cohort of 642 patients with advanced cirrhosis (defined as cirrhosis and MELD score ≥10) treated with a variety of DAA regimens, the overall SVR12 rate was 90.5%. Age <60, male sex, ascites, serum albumin <3.5 mg/dL, hepatocellular carcinoma, proton-pump inhibitor use, MELD score <16, and CTP class B/C were significantly associated with decreased odds of SVR12. In long-term follow-up at a median of 4 years after the end of treatment, a clinically meaningful decrease in MELD score of ≥3 occurred in 29% and a final MELD score of <10 was achieved in 25%. These data highlight that a proportion of patients with advanced cirrhosis who receive DAA therapy may not achieve significant long-term improvement in liver function (Verna, 2020). A recent retrospective study conducted in HCV-infected patients with decompensated cirrhosis found that DAA therapy was associated with reduced all-cause mortality and non-liver related deaths. In the 88% of patients who achieved SVR, the risk of mortality, hepatocellular carcinoma and liver transplantation was also reduced (Pageaux, 2022).
With the increased efficacy of DAAs in those with decompensated liver disease, a retrospective cohort study evaluated temporal trends, patient characteristics, and outcomes among adults with decompensated cirrhosis who were waitlisted for liver transplantation between January 1, 2005 and December 31, 2018. Overall, listing rates for HCV patients have decreased in the DAA era. However, delisting due to clinical improvement remains low, although such delisting has increased in more recent times (6.1% for 2013–2017; 5.2% for 2009–2012; 4% for 2005–2008; p <0.001). Ascites persisted in 48.6% and encephalopathy in 30.5% of patients at delisting, indicating that significant morbidity may persist in some patients over the long term, despite SVR (Bittermann, 2020).
Recommended regimens listed by pangenotypic, evidence level and alphabetically for:Patients With Decompensated Cirrhosisa Who Have Genotype 1-6 and Are Ribavirin Eligible |
||
|---|---|---|
| RECOMMENDED | DURATION |
RATING |
| Genotype 1-6: Daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg) with weight-based ribavirinb | 12 weeks | I, Ac |
| Genotype 1, 4, 5, or 6 only: Daily fixed-dose combination of ledipasvir (90 mg)/sofosbuvir (400 mg) with low initial dose of ribavirin (600 mg, increase as tolerated to weight-based dose) | 12 weeks | I, Ad |
|
a Includes CTP class B and class C patients who may or may not be candidates for liver transplantation, including those with hepatocellular carcinoma. b Low initial dose of ribavirin (600 mg) is recommended for patients with CTP class C cirrhosis; increase as tolerated. c Only available data for genotype 6 are in patients with compensated cirrhosis. d Only available data for genotypes 5 and 6 are in a small number of patients with compensated cirrhosis. |
||
Recommended regimens listed by pangenotypic, evidence level and alphabetically for:Patients With Decompensated Cirrhosisa Who Have Genotype 1-6 and Are Ribavirin Ineligible |
||
|---|---|---|
| RECOMMENDED | DURATION |
RATING |
| Genotype 1-6: Daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg) | 24 weeks | I, Ab |
| Genotype 1, 4, 5, or 6 only: Daily fixed-dose combination of ledipasvir (90 mg)/sofosbuvir (400 mg) | 24 weeks | I, Ac |
|
a Includes CTP class B and class C patients who may or may not be candidates for liver transplantation, including those with hepatocellular carcinoma. b Only available data for genotype 6 are in patients with compensated cirrhosis. c Only available data for genotypes 5 and 6 are in a small number of patients with compensated cirrhosis. |
||
Recommended regimens listed by pangenotypic, evidence level and alphabetically for:Patients With Decompensated Cirrhosisa and Genotype 1-6 Infection in Whom Prior Sofosbuvir- or NS5A Inhibitor-Based Treatment Failed |
||
|---|---|---|
| RECOMMENDED | DURATION |
RATING |
| Genotype 1-6: Daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg) with weight-based ribavirinb | 24 weeks | II, Cc |
| Prior sofosbuvir-based treatment failure, genotype 1, 4, 5, or 6 only: Daily fixed-dose combination of ledipasvir (90 mg)/sofosbuvir (400 mg) with low initial dose of ribavirin (600 mg; increase as tolerated) | 24 weeks | II, Cd |
|
a Includes CTP class B and class C patients who may or may not be candidates for liver transplantation, including those with hepatocellular carcinoma. |
||
Protease inhibitor-containing regimens (eg, glecaprevir, grazoprevir, paritaprevir, simeprevir, and voxilaprevir) are not recommended in patients with decompensated liver disease (see “Protease-Inhibitor Containing Regimens” discussion below for details).
The phase 3, open-label, multicenter, randomized ASTRAL-4 study enrolled 267 patients with genotype 1, 2, 3, 4, or 6 and decompensated cirrhosis (CTP class B at the time of screening) who were treatment naive (45%) or experienced (55%). Notably, 10% of patients were CTP class A or class C at treatment baseline. Patients were randomly assigned (1:1:1 ratio) to 12 weeks of a daily fixed-dose combination sofosbuvir (400 mg)/velpatasvir (100 mg); 12 weeks of sofosbuvir/velpatasvir plus weight-based ribavirin (1000 mg/d, weight <75 kg; 1200 mg/d, weight ≥75 kg); or 24 weeks of sofosbuvir/velpatasvir. Randomization was stratified by HCV genotype. All participants had a hemoglobin level >10 g/dL and an eGFR ≥50 mL/min (Curry, 2015b). The genotype/subtype distribution of the participants was 60% (159/267) genotype 1a; 18% (48/267) genotype 1b; 4% (12/267) genotype 2; 15% (39/267) genotype 3; 3% (8/267) genotype 4; and <1% (1/267) genotype 6. Ninety-five percent of patients had a baseline MELD score ≤15. SVR rates were 83% among those in the 12-week sofosbuvir/velpatasvir study arm, 94% in the 12-week sofosbuvir/velpatasvir plus ribavirin arm, and 86% in the 24-week sofosbuvir/velpatasvir arm. Among patients with genotype 1, the SVR rates were 88%, 96%, and 92%, respectively. Twenty-two participants had virologic failure, including 20 patients with relapse and 2 patients (genotype 3) with on-treatment virologic breakthrough. The presence of baseline NS5A resistant substitutions was not associated with virologic relapse. SVR rates among the 12 patients with CTP class B cirrhosis and genotype 2 were 100% (8/8) with sofosbuvir/velpatasvir for 12 weeks (with or without ribavirin), and 75% (3/4) with sofosbuvir/velpatasvir for 24 weeks. Among 39 patients with CTP class B cirrhosis with genotype 3, SVR rates were 50% (7/14) for 12 weeks of sofosbuvir/velpatasvir without ribavirin, 85% (11/13) for 12 weeks of sofosbuvir/velpatasvir plus ribavirin, and 50% (6/12) for 24 weeks of sofosbuvir/velpatasvir. Therefore, genotype 3 patients in particular appear to benefit from the addition of ribavirin to the regimen (Curry, 2015b). A recent real-world study investigated the safety and efficacy of sofosbuvir/velpatasvir with ribavirin in chronic genotype 1-6 HCV-related cirrhosis. All patients included were Childs-Pugh class B or C. After 12 weeks of treatment, of the 96% of patients who achieved SVR, 84.4% had improved Childs-Pugh scores and 64.6% had improved MELD scores. As such, the benefit of ribavirin therapy in addition to sofosbuvir/velpatasvir continues to be seen across all HCV genotypes (Liu, 2021). For patients with decompensated cirrhosis who are ribavirin ineligible, sofosbuvir/velpatasvir for 24 weeks is currently recommended, but additional studies involving larger numbers of patients are needed to define the optimal duration of therapy. At posttreatment week 12, 47% of patients had an improvement in CTP score, 42% had no change, and 11% had an increased CTP score. Nine patients (3%) died due to various causes during the study; no deaths were judged to be related to antiviral therapy. Serious adverse events were reported in 16% to 19% of the treated patients. Anemia (ie, hemoglobin <10 g/dL) was reported in 23% of the group receiving ribavirin, and 8% and 9% in those who received 12 weeks and 24 weeks of sofosbuvir/velpatasvir without ribavirin, respectively. Recently, the efficacy of sofosbuvir/velpatasvir therapy was studied in genotype 1 and 2 HCV-infected patients with decompensated cirrhosis. A small number of patients was treated for 12 weeks with this dual therapy. Therefore, a shorter therapy duration of 12 weeks may be sufficient for patients with decompensated cirrhosis who are ribavirin ineligible (Tada, 2021).
A real-world study that evaluated the safety and efficacy of sofosbuvir/velpatasvir (with or without ribavirin) demonstrated an SVR12 of 88% (intention-to-treat analysis) among patients with genotype 3 and decompensated cirrhosis; the treatment was noted to be safe (Wong, 2021). Sofosbuvir/velpatasvir has also been studied in a small number of patients with CTP class C cirrhosis. In a Japanese phase 3, open-label study of patients with CTP class B (77%) and CTP class C (20%) cirrhosis, 102 patients with genotype 1, 2, or 3 were randomized to 12 weeks of sofosbuvir/velpatasvir, with or without ribavirin (Takehara, 2019). Ribavirin dosing was weight based in CTP class B patients (600 mg/d ≤60 kg; 800 mg/d >60 to 80 kg; 1000 mg/d >80 kg) and 600 mg daily for all CTP class C patients. Overall SVR12 rates were 92% in each arm, but only 75% among patients with CTP class C cirrhosis.
There are no data on the outcomes of patients with decompensated cirrhosis and a history of prior sofosbuvir plus an NS5A inhibitor failure. However, in a phase 2, open-label, single-arm study using 24 weeks of sofosbuvir/velpatasvir plus weight-based ribavirin among patients with a history of treatment failure with an NS5A inhibitor-containing regimen, among 69 patients (28% with compensated cirrhosis) treated with sofosbuvir/velpatasvir plus ribavirin for 24 weeks, SVR rates were 97% for genotype 1 (83% with compensated cirrhosis), 93% for genotype 2 (no patients with cirrhosis), and 78% for genotype 3 (75% with compensated cirrhosis) (Gane, 2017). To date, there are no data for this regimen given for 24 weeks in patients with decompensated cirrhosis.
The phase 3, multicenter ASTRAL-1 trial evaluated the efficacy and safety of a 12-week course of daily fixed-dose sofosbuvir/velpatasvir among treatment-naive and -experienced patients with genotype 1, 2, 4, 5, or 6. The study included 35 patients with genotype 5 and 41 patients with genotype 6 (Feld, 2015). Overall SVR12 rates were 97% (34/35) in genotype 5 patients and 100% (41/41) in those with genotype 6. Of note, 100% SVR12 was achieved in the small number of genotype 5 patients (n=5) and genotype 6 patients (n=6) with compensated cirrhosis enrolled in ASTRAL-1.
The US-based, multicenter, randomized, open-label, phase 2 SOLAR-1 trial included 108 patients with genotype 1 or 4 and decompensated cirrhosis; 59 were categorized as CTP class B (score 7–9) and 49 were CTP class C (score 10–12). Participants were randomly assigned to 12 weeks or 24 weeks of the daily fixed-dose combination of ledipasvir (90 mg)/sofosbuvir (400 mg) plus ribavirin (initial dose of 600 mg, increased as tolerated) (Charlton, 2015b). After excluding the 7 patients who underwent liver transplantation during the study, SVR rates were 87% in CTP class B patients who received 12 weeks of treatment and 89% in those who received 24 weeks of treatment. Similarly, the SVR rates were 86% and 87%, respectively, with 12 weeks and 24 weeks of antiviral therapy in the CTP class C patients. Post-therapy virologic relapse occurred in 8% and 5% of the 12- and 24-week groups, respectively. In the majority of participants with CTP class B or C disease, the MELD and CTP scores decreased between baseline and posttreatment week 4. As expected, the frequency of serious adverse events increased with treatment duration in both the CTP class B group (10%, 12 weeks; 34%, 24 weeks) and the CTP class C group (26%, 12 weeks; 42%, 24 weeks). Most of the serious adverse events were related to ribavirin. The mean daily dose of ribavirin in the patients with decompensated cirrhosis was 600 mg. Therapy was discontinued in 7% of the CTP class B patients and 8% of the CTP class C patients in the 24-week treatment arm.
The multicenter (Europe, Canada, Australia, and New Zealand), randomized, open-label, phase 2 SOLAR-2 study included 160 patients with genotype 1 or 4 and decompensated cirrhosis (CTP class B or C). Study participants, who were treatment-naive or -experienced, were randomly assigned to 12 weeks or 24 weeks of daily fixed-dose ledipasvir (90 mg)/sofosbuvir (400 mg) plus ribavirin (initial dose of 600 mg, increased as tolerated). All participants had a hemoglobin level >10 g/dL and an estimated glomerular filtration rate (eGFR) >40 mL/min (Manns, 2016). Among the 150 patients with decompensated cirrhosis who completed therapy and had evaluable efficacy results, SVR12 was achieved in 85% (61/72) of those in the 12-week arm (90% [43/48] CTP class B; 75% [18/24] CTP class C). SVR 12 was achieved by 90% (70/78) of patients with decompensated cirrhosis in the 24-week study arm (98% [47/48] CTP class B; 77% [23/30] CTP class C). Post-therapy virologic relapse occurred in 6% (9/150) of the patients with decompensated cirrhosis who completed therapy (7 in 12-week arm; 2 in 24-week arm). Baseline CTP and MELD scores improved in the majority of the treated patients, but some participants experienced worsening hepatic function. Among nontransplanted patients whose MELD score was ≥15 at baseline, 80% (20/25) had a MELD score <15 at SVR12. Among those with a MELD score <15 at baseline, 4% (2/56) had a MELD score ≥15 at SVR12. During the study, 8% (13/160) of the enrolled patients with decompensated cirrhosis (2 CTP class B, 11 CTP class C) died from various causes but none of the deaths were attributed to antiviral therapy. Serious adverse events occurred in approximately 28% of patients with decompensated cirrhosis with no significant difference between the 12- and 24-week treatment arms.
A multicenter, double-blind study from France reported on the use of daily ledipasvir/sofosbuvir for 24 weeks compared to daily ledipasvir/sofosbuvir plus ribavirin for 12 weeks (with a 12-week placebo phase). Study participants included 154 patients with compensated cirrhosis and genotype 1 in whom prior peginterferon/ribavirin treatment failed (for most patients, treatment with peginterferon/ribavirin plus a protease inhibitor also failed) (Bourliere, 2015). The mean MELD score was 7 (range 6–16), 26% of patients had varices, and 13% had a low serum albumin level. The SVR12 rates were 96% with the 12-week regimen and 97% with the 24-week regimen. The most common adverse events were asthenia, headache, and pruritus. The frequency of severe adverse events and the need for early drug discontinuation were low in both treatment groups. In light of these results, it is reasonable to consider daily ledipasvir/sofosbuvir plus ribavirin for 12 weeks in patients with decompensated cirrhosis.
Collectively, the results from these trials indicate that a 12-week course of ledipasvir/sofosbuvir and ribavirin (initial dose of 600 mg, increased as tolerated) is an appropriate regimen for patients with decompensated cirrhosis and genotype 1 or 4. Such therapy may lead to objective improvements in hepatic function and reduce the likelihood of recurrent HCV infection after subsequent transplantation. Most patients received a ribavirin dose of 600 mg/d. Of 17 patients (16 genotype 1; 1 genotype 4) in the SOLAR-1 and SOLAR-2 trials (6 CPT class B; 11 CPT class C) who received ledipasvir/sofosbuvir plus ribavirin for 12 weeks or 24 weeks prior to or up to the time of liver transplantation, all had HCV RNA <15 IU/mL at the time of transplantation. Sixteen of the 17 patients achieved posttransplant SVR12; 1 patient died at postoperative day 15, but the HCV RNA was <15 IU/mL on day 14 (Yoshida, 2017).
Real-world cohort studies have reported SVR rates in patients with decompensated cirrhosis. Investigators from the United Kingdom reported on the use of 12 weeks of ledipasvir (90 mg)/sofosbuvir (400 mg) or daclatasvir (60 mg)/sofosbuvir (400 mg), with or without ribavirin, among 235 genotype 1 patients (Foster, 2016). SVR rates were similar in the participants receiving ledipasvir/sofosbuvir plus ribavirin or ledipasvir/sofosbuvir (86% and 81%, respectively). In this observational cohort study, 91% of the patients received ribavirin; only 6% discontinued ribavirin while 20% required a ribavirin dose reduction. MELD scores improved in 42% of treated patients and worsened in 11%. There were 14 deaths and 26% of the patients had a serious adverse event; none were treatment related.
The multicenter, prospective, observational HCV-TARGET study examined the real-world efficacy of ledipasvir/sofosbuvir (with or without ribavirin) for various treatment durations. SVR12 among genotype 1 patients with a history of clinically decompensated cirrhosis was 90% (263/293) among evaluable patients (Terrault, 2016). In this cohort, 29% of patients with decompensated cirrhosis were treated with ribavirin and 48% received 24 weeks of treatment.
A phase 2a, open-label study of 14 patients with compensated cirrhosis and genotype 1 in whom prior sofosbuvir-based therapy failed demonstrated that ledipasvir/sofosbuvir for 12 weeks was associated with a 100% SVR rate (Osinusi, 2014). In addition, results of an open-label, phase 2 study of 51 genotype 1 patients in whom prior sofosbuvir-based therapy failed demonstrated that a 12-week course of ledipasvir/sofosbuvir plus weight-based ribavirin (1000 mg/d to 1200 mg/d) led to an overall SVR12 of 98%, including 100% (14/14) among those patients with compensated cirrhosis (Wyles, 2015b).
Rarely, genotyping assays may indicate the presence of a mixed infection (eg, genotypes 1a and 2). Treatment data for mixed genotypes with DAAs are sparse but utilization of a pangenotypic regimen should be considered. When the correct drug combination or treatment duration is unclear, expert consultation should be sought.
Regimens not recommended for:
Patients With Decompensated Cirrhosis (Moderate or Severe Hepatic Impairment; Child-Turcotte-Pugh Class B or C)
|
|
|---|---|
| NOT RECOMMENDED |
RATING |
| Any protease inhibitor-containing regimen (eg, glecaprevir, grazoprevir, and voxilaprevir). | III, B |
| Interferon-based regimens | III, B |
The daily fixed dose combination of glecaprevir (300 mg)/pibrentasvir (120 mg) administered as three 100 mg/40 mg fixed-dose combination pills has not been studied in patients with decompensated cirrhosis and, pending additional safety data, is not recommended. In a retrospective analysis in a limited number of patients with portal hypertension, glecaprevir/pibrentasvir therapy for 8 or 12 weeks was equally efficacious in patients with and without features of portal hypertension. The therapy showed similar safety and tolerability features in both patient groups (Brown, 2022).
To date, the fixed-dose combination of elbasvir (50 mg)/grazoprevir (100 mg) has not been rigorously studied in patients with decompensated cirrhosis. A phase 2, nonrandomized, open-label study of elbasvir/grazoprevir (50 mg/50 mg) for 12 weeks was completed in 30 genotype 1 patients with CTP class B cirrhosis (Jacobson, 2019). SVR12 was 90% (27/30); 1 patient died of liver failure at posttreatment week 4 and 2 patients relapsed. At follow-up week 12, MELD scores improved in 41% (12/29) of treated patients, were unchanged in 38% (11/29), and worsened in 21% (6/29). However, there are no safety or efficacy data regarding the US Food and Drug Administration (FDA)-approved elbasvir/grazoprevir doses in patients with decompensated cirrhosis. Therefore, until further data are available, treatment of patients with decompensated cirrhosis with elbasvir/grazoprevir is not recommended.
Similarly, the daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg)/voxilaprevir (100 mg) has not been extensively studied in patients with hepatic decompensation. Thus, this regimen is not recommended for patients with decompensated cirrhosis (CTP class B or C) until further data are available. A recent real-world study conducted in a small number of patients with genotype 3 HCV infection and liver cirrhosis showed that the sofosbuvir/velpatasvir/voxilaprevir triple therapy was highly efficacious. However, poor tolerability was seen in patients with advanced liver disease (Papaluca, 2021). Similarly, a recent single-center retrospective case review study found this triple therapy to be highly efficacious, in patients with decompensated cirrhosis, when administered under careful observation to patients with a high likelihood of achieving SVR (Patel, 2021).
Interferon should not be given to patients with decompensated cirrhosis (moderate or severe hepatic impairment, CTP class B or C) because of the potential for worsening hepatic decompensation.
Recommended regimens listed by pangenotypic activity, evidence level and alphabetically for:Treatment-Naive and -Experienced Patients With Genotype 1-6 Infection in the Allograft Without Cirrhosis |
||
|---|---|---|
| RECOMMENDED | DURATION |
RATING |
| Daily fixed-dose combination of glecaprevir (300 mg)/pibrentasvir (120 mg)a | 12 weeks | I, B |
| Daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg) | 12 weeks | I, B |
| Genotype 1, 4, 5, or 6 only: Daily fixed-dose combination of ledipasvir (90 mg)/sofosbuvir (400 mg) | 12 weeks | I, B |
| a Dosing is 3 coformulated tablets (glecaprevir [100 mg]/pibrentasvir [40 mg]) taken once daily. Please refer to the prescribing information. | ||
Recommended regimens listed by pangenotypic, evidence level and alphabetically for:
Treatment-Naive and -Experienced Patients With Genotype 1-6 Infection in the Allograft With Compensated Cirrhosis
|
||
|---|---|---|
| RECOMMENDED | DURATION |
RATING |
| Daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg) | 12 weeks | I, B |
| Daily fixed-dose combination of glecaprevir (300 mg)/pibrentasvir (120 mg)a | 12 weeks | I, C |
| Genotype 1, 4, 5, or 6 only: Daily fixed-dose combination of ledipasvir (90 mg)/sofosbuvir (400 mg) | 12 weeks | I, A |
| a Dosing is 3 coformulated tablets (glecaprevir [100 mg]/pibrentasvir [40 mg]) taken once daily. Please refer to the prescribing information. | ||
Recommended regimens listed by pangenotypic, evidence level and alphabetically for:Treatment-Naive and -Experienced Patients With Genotype 1-6 Infection in the Allograft and Decompensated Cirrhosisa |
||
|---|---|---|
| RECOMMENDED | DURATION |
RATING |
| Daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg)/ ribavirin starting at 600 mg and increased as toleratedb | 12 to 24 weeksc | I, B |
| Genotype 1, 4, 5, or 6 only: Daily fixed-dose combination of ledipasvir (90 mg)/sofosbuvir (400 mg) with low initial dose of ribavirin (600 mg, increase as tolerated)b | 12 to 24 weeksc | I, B |
|
a Includes CTP class B and class C patients. b The starting dose of ribavirin should be 600 mg/d and increased or decreased as tolerated. If renal dysfunction is present, a lower starting dose is recommended. Maximum ribavirin dose is 1000 mg/d if <75 kg and 1200 mg/d if ≥75 kg body weight. c 24-week treatment duration is recommended if treatment experienced. |
||
Recommended regimen for:DAA-Experienced Patients With Genotype 1-6 Infection in the Allograft, With or Without Compensated Cirrhosisa |
||
|---|---|---|
| RECOMMENDED | DURATION |
RATING |
| Daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg)/voxilaprevir (100 mg)b | 12 weeks | I, C |
|
a Excludes CTP class B and class C patients. b For patients with cirrhosis plus multiple negative baseline characteristic, consideration should be given to adding ribavirin. The starting dose of ribavirin should be 600 mg/d and increased or decreased as tolerated. If renal dysfunction is present, a lower starting dose is recommended. Maximum ribavirin dose is 1000 mg/d if <75 kg and 1200 mg/d if ≥75 kg body weight. |
||
The MAGELLAN-2 trial was an open-label, multicenter, single-arm, phase 3 study that evaluated a 12-week course of the daily fixed-dose combination of glecaprevir (300 mg)/pibrentasvir (120 mg) administered as three 100 mg/40 mg fixed-dose combination pills in 80 liver transplant recipients and 20 kidney transplant recipients without cirrhosis. All genotypes were represented except genotype 5; 57% of participants had genotype 1 and 24% had genotype 3. Except for genotype 3 patients (all of whom were treatment naive), treatment-experienced patients were included (interferon or peginterferon ± ribavirin, or sofosbuvir plus ribavirin ± peginterferon). Eighty percent of patients had Metavir stage F0 or F1 fibrosis, 6% had F2, and 14% had F3. Cirrhotic patients were excluded. Any stable immunosuppressive regimen was allowed, except cyclosporine >100 mg/d and prednisone >10 mg/d. SVR was achieved in 98% (98/100) of patients with no virologic breakthroughs on treatment and 1 post-treatment relapse (Reau, 2018). There were no treatment discontinuations due to drug-associated adverse effects. One episode of mild rejection occurred that was assessed to be unrelated to drug interactions. A multicenter study from Japan treated 24 liver transplant recipients with recurrent HCV with 8 weeks or 12 weeks of glecaprevir/pibrentasvir (including 21% with F3/F4); 96% achieved SVR12. All 13 patients (genotype 1 or 2, without cirrhosis) treated for 8 weeks achieved SVR (Ueda, 2019). As data on the efficacy of glecaprevir/pibrentasvir in transplant recipients with cirrhosis and use of shorter treatment course (8 weeks versus 12 weeks) in those without cirrhosis are very limited, pending additional real-world data, a 12-week course is recommended regardless of stage. Additionally, for patients with cirrhosis plus other negative baseline factors, adding low-dose (600 mg) ribavirin may be a consideration.
The safety and efficacy of the fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg) for 12 weeks was evaluated in 79 (5 with cirrhosis, 4 DAA experienced) liver transplant recipients with genotype 1, 2, 3, or 4 (Agarwal, 2018). Treatment was well tolerated with 99% of patients completing treatment. Overall SVR12 rates by genotype were 93% genotype 1a (n=15); 96% genotype 1b (n=22); 100% genotype 2 (n=3); 97% genotype 3 (n=35); and 100% genotype 4 (n=4). Eighteen (23%) patients required a change in immunosuppression during treatment but none were for rejection or drug-drug-interactions. Most patients were on calcineurin inhibitor-based immunosuppression (71% on tacrolimus, 14% on cyclosporine).
In the nontransplant setting (discussed in detail in the Initial and Retreatment sections), the phase 3, double-blind, placebo-controlled ASTRAL-1 study reported on 742 treatment-naive or -experienced patients with genotype 1, 2, 4, 5, or 6 who were randomly assigned in a 5:1 ratio to sofosbuvir/velpatasvir or placebo for 12 weeks (Feld, 2015). All patients with genotype 5 (n=35) received active treatment. Thirty-two percent (201/624) of patients randomized to active therapy were treatment experienced and 19% (121/624) had compensated cirrhosis (CTP class A). The genotype distribution in the active treatment arm was 34% (n=210) genotype 1a; 19% (n=118) genotype 1b; 17% (n=104) genotype 2; 19% (n=116) genotype 4; 6% (n=35) genotype 5; and 7% (n=41) genotype 6. The overall SVR was 99% (95% CI, 98 to >99). The side effect/adverse event profile of sofosbuvir/velpatasvir was similar to placebo.
In the phase 3, open-label ASTRAL-3 study, 552 treatment-naive or -experienced patients with genotype 3 (with or without compensated cirrhosis) were randomized in a 1:1 ratio to 12 weeks of sofosbuvir/velpatasvir or 24 weeks of sofosbuvir plus weight-based ribavirin. SVR12 was 95% (95% CI, 92 to 98) for the sofosbuvir/velpatasvir treatment arm, which was superior to the SVR12 of 80% (95% CI, 75 to 85) for patients receiving sofosbuvir plus ribavirin for 24 weeks (Foster, 2015a).
The phase 3, open-label ASTRAL-4 study enrolled 267 treatment-naive or -experienced (55%) patients with genotype 1, 2, 3, 4, or 6 and decompensated cirrhosis (CTP class B at the time of screening). Patients were randomized in a 1:1:1 ratio to 12 weeks of sofosbuvir/velpatasvir, 12 weeks of sofosbuvir/velpatasvir plus weight-based ribavirin, or 24 weeks of sofosbuvir/velpatasvir. SVR12 rates were 83% (75/90) for the 12-week sofosbuvir/velpatasvir regimen, 94% (82/87) for the 12-week sofosbuvir/velpatasvir plus ribavirin regimen, and 86% (77/90) for the 24-week sofosbuvir/velpatasvir regimen (Curry, 2015b). Among patients with genotype 1, SVR12 rates were 88% and 96% with 12 weeks of sofosbuvir/velpatasvir without and with ribavirin respectively, and 92% with sofosbuvir/velpatasvir for 24 weeks. Virologic relapse occurred in 12% and 9% of patients in the 12-week and 24-week sofosbuvir/velpatasvir arms, respectively, compared to 2% in the 12-week sofosbuvir/velpatasvir plus ribavirin study arm. Although the ASTRAL-4 study was not powered to generate statistical significance, these results suggest that sofosbuvir/velpatasvir with ribavirin for 12 weeks is the optimal choice for patients with genotype 1 or 3 who have decompensated cirrhosis. The participant numbers were too small for genotypes 2, 4, and 6 to differentiate the comparative efficacy of the treatment arms. Reflecting the approach in nontransplant patients with decompensated cirrhosis, liver transplant recipients with hepatic decompensation are recommended to receive sofosbuvir/velpatasvir plus ribavirin for 12 to 24 weeks, depending upon presence of other negative prognostic features at baseline (ie, treatment experienced, genotype 3, presence of hepatocellular carcinoma).
Velpatasvir is a substrate for CYP3A4, CYP2C8, and CYP2B6, a weak inhibitor of P-gp and OATP transporters, and a moderate inhibitor of the breast cancer resistance protein (BCRP) membrane transporter. As such, velpatasvir is moderately affected by potent inhibitors and, to a greater extent, potent inducers of enzyme/drug transporter systems (Mogalian, 2016). Based on this profile, which is similar to ledipasvir, clinically significant drug-drug interactions would not be expected for coadministration of sofosbuvir/velpatasvir with common immunosuppressive agents (eg, tacrolimus, cyclosporine, corticosteroids, mycophenolate mofetil, or everolimus).
The SOLAR-1 study was a large, US-based, multicenter, open-label, phase 2 trial that included 223 liver transplant recipients with genotype 1 or 4 whose baseline characteristics encompassed a broad spectrum of histologic and clinical severity of HCV recurrence. One hundred and eleven patients were Metavir stage F0 to F3, 51 had compensated CTP class A cirrhosis, and 61 had decompensated CTP class B or class C cirrhosis. Study participants were randomly assigned to 12 weeks or 24 weeks of a fixed-dose combination of ledipasvir (90 mg)/sofosbuvir (400 mg) plus ribavirin. The ribavirin dose was weight based for patients without cirrhosis or with compensated cirrhosis (1000 mg/d [<75 kg] to 1200 mg/d [≥ 75 kg]). For patients with CTP class B or class C cirrhosis, ribavirin was initiated at 600 mg/d followed by dose escalation as tolerated. Only 4% of enrolled participants discontinued treatment prematurely because of adverse events related to the study drugs (Charlton, 2015b). On an intention-to-treat basis, SVR was achieved in 96% (53/55) and 98% (55/56) of liver transplant patients without cirrhosis in the 12- and 24-week treatment arms, respectively. Among those with compensated cirrhosis, SVR was 96% in both the 12- and 24-week treatment arms. Efficacy was lower in patients with CTP class B or class C cirrhosis post liver transplantation. Among those with CTP class B cirrhosis, SVR rates were 86% and 88% in the 12- and 24-week treatment arms, respectively. Among patients with CTP class C cirrhosis, SVR rates were 60% and 75% in the 12- and 24-week treatment arms, respectively. Mortality rate during the study was 10% among patients with CTP class B or class C cirrhosis (Charlton, 2015b).
Similar results were achieved using an identical study design in the SOLAR-2 study, which was conducted in Europe, Australia, Canada, and New Zealand. The study included 168 liver transplant recipients with genotype 1 or 4 infection. Among the post-transplantation patients, 101 had no cirrhosis (Metavir stage F0 to F3), 67 had CTP class A compensated cirrhosis, 45 had CTP class B cirrhosis, and 8 had CTP class C decompensation. SVR rates in post-transplantation, noncirrhotic patients were 94% (49/52) and 100% (49/49) for 12 weeks and 24 weeks of treatment, respectively. Among patients with compensated cirrhosis after transplantation, SVR was 97% (33/34; 32/33) in both the 12- and 24-week treatment arms. For patients with CTP class B cirrhosis, comparable SVR rates were 95% (21/22) and 100% (23/23), respectively. Among those with CTP class C cirrhosis, SVR rates were 33% (1/3) and 80% (4/5), respectively. Considering both pre- and post-transplantation patients with CTP class B or class C cirrhosis, SVR rates were 85% (61/72) and 90% (70/78) for 12 weeks and 24 weeks of treatment, respectively.
An observational HCV-TARGET cohort study provides real-world data based on experience with 347 liver, 60 kidney, and 36 dual liver and kidney transplant recipients. Among the enrolled patients, 86% had genotype 1, 44% had cirrhosis, 26% had a history of liver decompensation, and 54% had a prior treatment failure with a non-NS5A inhibitor regimen (Saxena, 2017). Among the 279 participants treated with ledipasvir/sofosbuvir for 12 weeks or 24 weeks, the SVR rates were 97% (152/157) for those also taking ribavirin and 95% (116/122) for patients not taking ribavirin. Patients who received ribavirin were more frequently genotype 1a (versus genotype 1b), treatment experienced, and without renal dysfunction. The rate of therapy discontinuation due to an adverse event was 1.3%, highlighting the safety of the drug combination. Acute graft rejection occurred during or after cessation of therapy in 1.4% (6/415) of patients. These episodes were not judged to be a direct consequence of the antiviral regimen but serve to remind clinicians of the need to monitor immunosuppressive agent levels during direct-acting antiviral (DAA) therapy.
Another multicenter cohort of 162 patients (98% genotype 1) assessed treatment with ledipasvir/sofosbuvir (with or without ribavirin) for 8 weeks, 12 weeks, or 24 weeks. Duration of treatment and ribavirin use were provider determined. Overall SVR12 rates were 94% and 98% in those treated with ledipasvir/sofosbuvir without or with ribavirin, respectively (Kwok, 2016). SVR12 rates in patients treated for 8 weeks, 12 weeks, or 24 weeks with the ribavirin-free regimen were 86% (6/7), 94% (65/69), and 95% (39/41), respectively. SVR12 rates in the ribavirin-inclusive groups were 97% (38/39) and 100% (6/6) for 12 weeks and 24 weeks of treatment, respectively.
The multicenter ANRS CO23 CUPILT study investigators reported their experience with sofosbuvir plus an NS5A inhibitor (daclatasvir or ledipasvir ± ribavirin) among 512 liver transplant recipients with recurrent HCV who met inclusion criteria for analysis (Houssel-Debry, 2018). The genotype distribution of the participants was 70% (n=359) genotype 1, 1% (n=7) genotype 2, 18% (n=93) genotype 3, 10% (n=50) genotype 4, and <1% (n=3) genotype 5. Twenty-one percent had cirrhosis and 34% had prior treatment experience. The regimens and treatment durations were sofosbuvir plus an NS5A inhibitor without ribavirin for 12 weeks (n=156) or 24 weeks (n=239), and sofosbuvir plus an NS5A inhibitor and ribavirin for 12 weeks (n=47) or 24 weeks (n=70). SVR12 rates were 94%, 99%, 96%, and 93%, respectively. Twenty patients experienced treatment failure and in a multivariate analysis, fibrosis stage, prior treatment, genotype, and baseline HCV viral load did not adversely impact SVR12 rates in the 4 treatment groups. The investigators concluded that 12 weeks of sofosbuvir plus an NS5A inhibitor without ribavirin was an effective regimen regardless of fibrosis stage, genotype, and prior treatment experience.
Collectively, these real-world experiences indicate high SVR rates can be attained without inclusion of ribavirin in liver transplant patients. However, all factors leading clinicians to include or exclude ribavirin cannot be discerned from these observational studies. The safest presumption is that ribavirin may contribute to the high SVR rates and be relevant for patients with unfavorable baseline characteristics (eg, cirrhosis, prior treatment experience). Thus, ribavirin-free therapy is recommended for patients with a favorable baseline profile and ribavirin-inclusive therapy is recommended for those with an unfavorable baseline profile.
Most clinical trials to date have focused on patients who were at least 6 months post transplantation, but there is no a priori reason not to consider earlier treatment if the patient is on stable immunosuppression and has recovered from postoperative complications. Treatment during the first 6 to 12 months post transplantation certainly seems reasonable to reduce the likelihood of treating patients with more advanced liver disease. A phase 2 study of prophylactic ledipasvir/sofosbuvir enrolled 16 genotype 1 liver transplant recipients (most with hepatocellular carcinoma as the indication). Treatment was initiated immediately preoperatively and continued for 4 weeks post transplantation (Levitsky, 2016). SVR12 post transplantation was attained in 88% (15/16) of patients. While these results are too preliminary upon which to base recommendations, the findings provide additional data on the safety of ledipasvir/sofosbuvir early in the post-transplantation period.
There is limited experience with sofosbuvir/velpatasvir/voxilaprevir in liver transplant recipients. In a single case report of a prior DAA regimen failure, successful treatment of recurrent HCV after liver transplant with sofosbuvir/velpatasvir/voxilaprevir was achieved (Cardona-Gonzalez, 2018). The patient had genotype 3 infection and acute hepatitis post liver transplant. He was treated with sofosbuvir/velpatasvir/voxilaprevir for 16 weeks with ribavirin added during the last 8 weeks of therapy. In a subsequent case series, 6 liver transplant recipients with HCV genotype 1 ± genotype 4 infection who had a previous DAA treatment failure were treated with sofosbuvir/velpatasvir/voxilaprevir. Participants received a 12-week course of therapy and all achieved SVR. Minor reductions in calcineurin inhibitor dosing were required but no adverse events or rejection episodes were reported (Higley, 2020).
Rarely, genotyping assays may indicate the presence of a mixed infection (eg, genotypes 1a and 2). Treatment data for mixed genotypes with DAAs are sparse but utilization of a pangenotypic regimen should be considered. When the correct combination or treatment duration is unclear, expert consultation should be sought.
The interactions of DAA agents and calcineurin inhibitors are complex and unpredictable without formal studies of drug-drug interactions. A summary of drug-drug interactions between calcineurin inhibitors and DAAs with recommended dosing is provided in the table below. Based on the metabolism of grazoprevir and elbasvir, a 15-fold increase in grazoprevir AUC and a 2-fold increase in elbasvir AUC can be expected with cyclosporine coadministration. Therefore, this combination should be avoided. Since a 40% to 50% increase in tacrolimus level is predicted during coadministration with grazoprevir, no dosing adjustments are anticipated but tacrolimus levels should be monitored.
Table. DAA Interactions With Calcineurin Inhibitors
Cyclosporine (CSA) |
Tacrolimus (TAC) |
|
|---|---|---|
| Sofosbuvir (SOF) | 4.5-fold ↑ in SOF AUC, but GS-331007 metabolite unchanged; no a priori dose adjustment | No interaction observed; no a priori dose adjustment |
| Ledipasvir | No data; no a priori dose adjustment | No data; no a priori dose adjustment |
| Elbasvir / grazoprevir (EBR/GZR) | 15-fold ↑ in GZR AUC and 2-fold ↑ in EBR AUC; combination is not recommended | 43% ↑ in TAC; no a priori dose adjustment |
| Velpatasvir | No interaction observed; no a priori dose adjustment | No data; no a priori dose adjustment |
| Glecaprevir / pibrentasvir (GLE/PIB) | 5-fold ↑ in GLE AUC with higher doses (400 mg) of CSA; not recommended in patients requiring stable CSA doses >100 mg/day | 1.45-fold ↑ in TAC AUC; no a priori dose adjustment; monitor TAC levels and titrate TAC dose as needed |
| Sofosbuvir / velpatasvir / voxilaprevir (SOF/VEL/VOX) | 9.4-fold ↑ in VOX AUC; combination is not recommended | No data; no a priori dose adjustment |
| AUC=area under the curve | ||
With the large disparity between patients in need of organ transplantation and available donor organs, many transplant programs are turning to the use of organs from HCV-viremic donors. In the past, organs from HCV-viremic donors were primarily used in recipients with chronic hepatitis C or discarded. With the advent of safe and effective HCV DAA regimens, however, organs from HCV-viremic donors may be considered for use in recipients without HCV infection. Use of these organs increases the pool of available organs, patient access to transplantation (Sageshima, 2018), and potentially reduces waitlist time (Bhamidimarri, 2017); (Scalea, 2015) and related mortality (Sawinski, 2019); (Shelton, 2018); (Kucirka, 2012); (Altshuler, 2022).
All organ donors undergo HCV-antibody and HCV nucleic acid testing (NAT). Nonhepatic donors who are HCV antibody positive but HCV RNA negative likely pose a negligible risk of HCV transmission to the recipient, although more data are needed to confirm this. However, among increased-risk donors (as defined by the US Public Health Service [PHS] guidelines) who had a recent HCV exposure, HCV RNA may not yet be detectable and transplant recipients from these donors should be monitored for HCV in addition to HBV and HIV per the increased-risk donor testing protocols (Levitsky, 2017); (Seem, 2013b). Transplant recipients who receive a liver from an HCV-antibody–positive/HCV-RNA–negative donor should be monitored more closely after transplantation given the potential risk for HCV transmission (Bari, 2018); (Sobotka, 2021). Donors who are HCV RNA positive (with or without anti-HCV) pose the highest risk for HCV transmission to transplant recipients. Because of the significant risk for HCV infection when transplanting an organ from an HCV-viremic donor into an HCV-uninfected recipient, rigorous informed consent, including discussion of potential secondary risks to caregivers from needlestick exposures (Kim, 2022), and post-transplantation, HCV-related follow-up processes are recommended.
Recommendations When Considering Use of HCV-Viremic Donor Organs in HCV-Uninfected Recipients |
|
|---|---|
| RECOMMENDED |
RATING |
|
Informed consent should include the following elements:
|
I, C |
Transplant programs should have a programmatic strategy to:
|
I, C |
Recent data indicate increasing acceptance of organs from HCV-viremic donors among HCV-uninfected recipients (Cotter, 2019); (Potluri, 2019); (Bowring, 2018). Although no published data are available regarding the long-term (beyond 1 to 2 years) consequences to HCV-negative recipients transplanted with organs from HCV-viremic donors who are treated post-transplant with DAAs, limited short-term data from liver, kidney, heart, and lung transplant programs are encouraging.
Among 10 HCV-negative liver transplant recipients of organs from HCV-viremic donors, 100% achieved SVR12 with 12 to 24 weeks of various DAA regimens (Kwong, 2019). The median time from transplantation to treatment initiation was 43 days (interquartile range [IQR] 20-59 days). Noteworthy was the high rate of acute cellular or antibody-mediated rejection (30%) during or after DAA therapy in this study. In another study of 14 HCV-negative liver transplant recipients from HCV-viremic donors, treated with glecaprevir/pibrentasvir for 12 weeks starting within 5 days of transplant, SVR rates were 100% and only one patient experienced acute rejection (Bethea, 2020). In another single center experience, 61 HCV-negative recipients of liver allografts from HCV-viremic donors were compared to 231 HCV-negative recipients of liver allografts from HCV-negative donors (Bohorquez, 2021a). Of the 61 patients in the study group, 56 received antiviral therapy; treatment was initiated a median of 66.9 days following transplantation. Four study group participants died (within 1 year following liver transplantation), one was persistently aviremic, and another experienced a complex post-operative course). Of the 51 patients with complete treatment data, 64% were treated with glecaprevir/pibrentasvir and 36% received sofosbuvir/velpatasvir. All patients achieved SVR12; one participant required retreatment with sofosbuvir/velpatasvir/voxilaprevir after relapse. There were no significant differences between recipients of allografts from HCV-viremic vs HCV-negative donors in terms of other clinical outcomes such as acute cellular rejection, kidney dysfunction, or survival. A retrospective study of deceased donor liver transplantations in the US from January 2008 through January 2018 demonstrated that 2-year graft survival was similar, regardless of HCV status concordance or discordance between the allograft donor and recipient (Cotter, 2019). In a single-center retrospective study of 21 HCV-seronegative recipients who received a liver transplant from HCV-viremic donors, 20 (95%) of recipients had confirmed HCV viremia and 100% of the 15 patients with available data achieved SVR12 after DAA treatment. There were equivalent rates of post-transplant complications between the 21 recipients who received a liver from HCV-viremic donors when compared to 21 recipients who received a liver from HCV antibody positive / NAT negative donors (Sobotka, 2021).
In a prospective, multicenter (n=6), single-arm, open-label clinical trial, 13 HCV-negative liver transplant recipients received allografts from HCV-viremic donors. Participants were treated with 12 weeks of sofosbuvir/velpatasvir; the median time from transplantation to antiviral therapy was 7 days (Terrault, 2020). All liver transplant recipients achieved SVR12. Serious adverse events possibly related to study participation among the liver recipients included antibody mediated rejection, biliary sclerosis, cardiomyopathy, and graft-versus-host disease (which eventually led to the patient’s death). In a prospective multicenter (n=3) observational study, 20 HCV-negative patients received a liver transplant from HCV-viremic donors and all recipients had HCV viremia confirmed within 3 days post-transplant and achieved SVR12 after receiving DAA treatment (median 27.5 days post-transplant) (Aqel, 2021). One patient who was started on DAA treatment on post-op day 24 developed end-stage renal disease secondary to HCV-related acute membranous nephropathy and died 14 months post-transplant due to septic shock from a presumed infection.
Unlike with other organs, shorter durations of HCV therapy should not be used in recipients of livers from HCV-viremic donors because of the large reservoir of HCV in the transplanted organ. Additionally, although prophylactic or pre-emptive therapy has not been as strongly stressed for recipients of liver grafts from HCV-viremic donors, a case report noted the development of acute kidney injury (with proteinuria) in the first month posttransplant due to HCV-associated focal proliferative glomerulonephritis. This case report highlights the potential for HCV-related, extrahepatic manifestations in the early posttransplant setting (Bohorquez, 2021b). The prospective multicenter noted above (Aqel, 2021) also highlights the importance of considering the initiation of DAA treatment earlier post-transplant given that one liver transplant recipient had biopsy-proven acute HCV-related glomerulonephritis on post-op day 18, which was 6 days prior to the initiation of DAA treatment, and went on to develop end stage renal disease despite having achieved SVR12. This patient died due to presumed infectious complications. The possible high risk for immunologic complications (eg, rejection) in liver recipients from HCV-viremic donors treated with DAA therapy requires further study but vigilance is appropriate.
Recommendation Regarding Timing of DAA Therapy for HCV-Negative Recipients of HCV-Viremic Liver Transplant |
|
|---|---|
| RECOMMENDED |
RATING |
| Earlya treatment with a pangenotypic DAA regimen is recommended when the patient is clinically stable. | II, B |
| a Early treatment refers to starting within the first 2 weeks after liver transplant but preferably within the first week when the patient is clinically stable. | |
|
Recommended regimens listed by pangenotypic, evidence level and alphabetically for: Treatment of HCV-Uninfected Recipients of Liver Grafts from HCV-Viremic Donors |
||
|---|---|---|
| RECOMMENDED | DURATION |
RATING |
| Daily fixed-dose combination of glecaprevir (300 mg)/pibrentasvir (120 mg)b | 12 weeks | I, C |
| Daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg) | 12 weeks | I, C |
a Other considerations in selection of the DAA regimen:
b Dosing is 3 coformulated tablets (glecaprevir [100 mg]/pibrentasvir [40 mg]) taken once daily. Please refer to the prescribing information. |
||
In the THINKER trial, 10 HCV-uninfected kidney transplant recipients received allografts from genotype 1 HCV-viremic donors and were treated with 12 weeks of elbasvir/grazoprevir; 100% achieved SVR (Goldberg, 2017). In a 1-year follow-up study that included 10 additional participants (n=20) who received 12 to 16 weeks of elbasvir/grazoprevir (± ribavirin), all achieved SVR12. Kidney function in those who received kidneys from HCV-infected donors was comparable to matched controls who received allografts from HCV-uninfected donors (Reese, 2018). A separate open-label trial similarly demonstrated 100% SVR12 with 12 weeks of elbasvir/grazoprevir (± sofosbuvir) therapy initiated immediately prior to transplantation in 10 HCV-uninfected kidney transplant recipients of allografts from HCV-viremic donors (Durand, 2018). Notably, organ recipients in this study received the first dose of elbasvir/grazoprevir on call to the operating room. Of the 10 patients treated, only 3 had detectable HCV viremia compared to 100% in the THINKER trial, which utilized the same regimen but initiated therapy on day 3 after transplantation.
In a prospective, multicenter (n=7) study to transplant hepatitis C-infected kidneys (ie, the MYTHIC trial), 30 HCV-negative recipients received kidney allografts from HCV-viremic donors. Early initiation of glecaprevir/pibrentasvir (target was within 3 days posttransplant) for 8 weeks resulted in 100% SVR12; there were no significant treatment-related adverse events (Sise, 2020). Three episodes of acute rejection were noted but all patients had good graft function at 6 months follow-up. Three patients developed transient BK viremia and 4 (40%) of the 10 recipients who were CMV donor seropositive, CMV recipient seronegative developed CMV disease within the first-year post-transplant. One-year survival was 93% and 1-year graft function was excellent (median creatinine 1.17; IQR: 1.02-1.38 mg/dl) (Sise, 2021).
A prospective, multicenter, single-arm, open-label clinical trial evaluated the safety and efficacy 12 weeks of sofosbuvir/velpatasvir among 11 HCV-negative kidney transplant recipients who received grafts from HCV-viremic donors (Terrault, 2020). The median time from transplant to initiation of DAA therapy was 16.5 days; all kidney transplant recipients in this study achieved SVR12. No serious adverse events related to study participation were noted in the kidney recipients in this study. The REHANNA trial evaluated a shortened 4-week course of glecaprevir/pibrentasvir treated (compared to the standard 8 weeks) among HCV-negative kidney transplant recipients who received grafts from HCV-viremic donors. The first dose was administered prior to organ perfusion. All 10 patients achieved SVR12 and there were no adverse outcomes noted (Durand, 2021). Other studies in HCV discordant kidney donors and transplant recipients have also demonstrated high SVR12 rates without any treatment-related toxicities (Franco, 2019); (Friebus-Kardash, 2019). A single-center, retrospective cohort study compared 1-year outcomes for 65 transplant recipients who received a kidney from HCV viremic donors to 59 recipients who received a kidney from HCV negative donors (Molnar, 2021). Allograft biopsy findings and kidney allograft function during the first-year post-kidney transplantation were assessed and there were no statistically significant differences between the HCV positive and HCV negative cohorts with regards to delayed graft function rates, estimated glomerular filtration rates (eGFR), and proportions of patients with cellular rejection, antibody mediated rejection, or de novo DSAs.
A study of HCV-uninfected recipients who received a heart transplant from an HCV-viremic donor showed that using a 12-week course of elbasvir/grazoprevir initiated a few days after transplantation (once the recipient became viremic) resulted in SVR12 in 9 out of the 10 evaluable patients (McLean, 2019). In the DONATE HCV trial, 44 HCV-uninfected lung (n=36) and heart transplant (n=8) recipients from HCV-viremic donors sofosbuvir/velpatasvir was administered prophylactically or preemptively, starting within a few hours after transplantation and continued for 4 weeks (compared to the standard 12-week course). Among the initial 35 patients with at least 6 months of follow-up after transplantation, 100% achieved SVR and had excellent graft function (Woolley, 2019). There was an increase in the proportion of the HCV-viremic lung cohort who had acute cellular rejection compared to the non-HCV lung cohort, although this finding was not statistically significant and longer-term follow-up is needed to assess for chronic rejection. In a study of 20 HCV-uninfected heart transplant recipients of allografts from HCV-viremic donors, patients were treated prophylactically or preemptively with glecaprevir/pibrentasvir beginning just prior to transplantation and continued for 8 weeks. All participants achieved SVR12, and patient and graft survival were 100% with a median follow-up of 10.7 months (Bethea, 2019). Another clinical trial evaluated 22 HCV-uninfected lung transplant recipients of allografts from HCV-viremic donors; the 20 patients who became viremic after transplantation were treated with 12 weeks of sofosbuvir/velpatasvir beginning 2 to 6 weeks after transplantation (median 21 days; IQR 16.76-24.75 days). All lungs from HCV-viremic donors were treated with ex-vivo lung perfusion ± ultraviolet C perfusate irradiation to reduce HCV RNA concentration and infectivity, likely contributing to a slower rise in HCV viral load among recipients. Although all 20 DAA-treated patients had undetectable HCV RNA at the end of treatment, 2 patients experienced post-treatment relapse. One patient experienced severe hepatitis with early signs of fibrosing cholestatic hepatitis [FCH] on liver biopsy, and both patients exhibited complex NS3A and NS5A RASs at relapse. Both relapsed patients were successfully retreated with 24 weeks of sofosbuvir/velpatasvir/voxilaprevir plus ribavirin and achieved SVR12 (Cypel, 2019).
A study of 22 heart transplants from HCV-viremic donors evaluated an 8-week course of glecaprevir/pibrentasvir initiated 6–11 days after transplantation, once the viremia developed. Two patients had DAA interruptions. No differences were noted between the HCV-viremic vs HCV-aviremic donor cohorts in terms of survival or rejection (Reyentovich, 2020). Another study evaluated 38 thoracic organ transplants (22 heart; 16 lung) from HCV-viremic donors. Treatment with glecaprevir/pibrentasvir was initiated at the time of detectable viremia (mean 7 days) among the heart recipients and within 3 days after transplantation for the lung recipients; all participants achieved SVR12 (Smith, 2021). DAA treatment interruption occurred in 2 patients due to hyperbilirubinemia. One patient resumed treatment within a few days; the other patient’s treatment course was shortened to 10 days. Both patients still achieved SVR12. In the heart transplant recipients, all patients became viremic within the first week after transplantation. In contrast, only 11 of the 16 lung transplant recipients developed viremia. Overall, investigators noted reduced HCV transmission, lower viral loads, and more rapid clearance in the lung transplant patients who received earlier treatment (Smith, 2021). In both of these studies, initiation of treatment within a few days after transplantation was associated with an occasional need for treatment interruption, although all recipients still achieved SVR12 (Reyentovich, 2020); (Smith, 2021).
A separate study conducted among 50 heart transplant recipients (22 received hearts from HCV-viremic donors), an 8-week course of glecaprevir/pibrentasvir was initiated once viremia developed (mean 7.2 days) (Gidea, 2020). Investigators noted a higher proportion of acute cellular rejection in the HCV-viremic vs HCV-aviremic donor study groups (14/22 vs 5/28, respectively; p=0.001) in the first 2 months and at 180 days (17/22 vs 12/28, respectively; p=0.02). These findings raise concern about a potential association between HCV-viremic donors and rejection.
While these early results are encouraging, the overall number of published cases is small and treatment approaches notably variable. Known reported risks include DAA treatment failure with emergence of complex RASs and possible severe or rapidly progressive liver disease (fibrosing cholestatic hepatitis) (Cypel, 2019); (Kapila, 2019); (Molnar, 2019). Additionally, ethical and scientific issues remain, including avoidance of selection bias, optimal timing of DAA therapy, and long-term graft and patient outcomes. Due to the limited and heterogeneous experience and lack of longer-term safety data, strong consideration should be given to performing these transplantations with a rigorous informed consent process as recommended by the American Society of Transplantation consensus panel (Levitsky, 2017).
In addition, there have been an increasing number of dual organ transplants performed from HCV-viremic donors for heart-kidney recipients nationally between August 2015 and August 2020. Analyses from the UNOS registry demonstrated similar 1-year survival between 90 HCV donor seropositive and 896 HCV donor seronegative heart-kidney recipients using unadjusted and adjusted Cox-proportional hazards-regression models including in propensity-score matched cohorts (Madan, 2021); (Diaz-Castrillon, 2022).
Recommendation Regarding Timing of DAA Therapy for HCV-Negative Recipients of HCV-Viremic Non-Liver Solid Organ Transplant |
|
|---|---|
| RECOMMENDED |
RATING |
| Prophylactica or preemptiveb treatment with a pangenotypic DAA regimen is recommended. | II, B |
|
a Initiate DAA therapy immediately pretransplant or on day 0 posttransplant. No HCV RNA testing of the transplant recipient is required |
|
|
Recommended regimens listed by pangenotypic, evidence level and alphabetically for: Treatment of HCV-Uninfected Recipients of Non-Liver Organs from HCV-Viremic Donors |
||
|---|---|---|
| RECOMMENDED | DURATION |
RATING |
| Daily fixed-dose combination of glecaprevir (300 mg)/pibrentasvir (120 mg)b | 8 weeks | I, C |
| Daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg) | 12 weeks | I, C |
a Other considerations in selection of the DAA regimen:
b 8 weeks is recommended for prophylactic/preemptive treatment approaches. However, if treatment initiation is delayed beyond the first week after transplant, treatment should be continued for 12 weeks. Dosing is 3 coformulated tablets (glecaprevir [100 mg]/pibrentasvir [40 mg]) taken once daily. Please refer to the prescribing information. |
||
Initiation of DAA therapy for HCV-negative recipients of a non-liver allograft from an HCV-viremic donor can occur prophylactically/preemptively (ie, perioperatively without confirmation of viremia in the recipient) or reactively after documentation of HCV viremia. The goal is to undertake DAA therapy as early as clinically possible to minimize the duration of HCV viremia in the recipient and avoid the development of acute hepatitis and other non-hepatic complications of HCV infection. Initiating prophylactic/preemptive DAA therapy before viremia occurs may reduce the likelihood of complications, such as FCH, acute HCV-related glomerulonephritis, acute pancreatitis, acute cellular rejection, and allograft vasculopathy (Gidea, 2020); (Schlendorf, 2020); (Bethea, 2019); (Cypel, 2019); (Kapila, 2019); (Woolley, 2019); (Molnar, 2019); (Durand, 2018). A prophylactic/preemptive treatment approach may also allow for a shorter duration of DAA therapy in non-liver transplant recipients of organs from HCV-viremic donors (Woolley, 2019). A recent trial evaluated the use glecaprevir/pibrentasvir combined with ezetimibe 10 mg (as an inhibitor of HCV entry) in 30 recipients of nonhepatic organs (lung, heart, kidney) from HCV-viremic donors. The drugs were administered with 1 dose before and for 7 days after transplantation. With this short therapy, none of the 30 individuals developed chronic HCV infection. It is unknown if infection occurred and was rapidly cleared or if it was prevented entirely (Feld, 2020). Although intriguing, short duration approaches are not currently recommended outside of a clinical trial setting and have only been studied in the context of non-liver transplantation.
Though initiating HCV treatment as early as possible post-transplant may be clinically beneficial, barriers to initiating DAA treatment prophylactically/preemptively include the cost of DAA treatment and protracted insurance authorizations. One study compared the clinical and financial impact between an institution-subsidized course of initial DAA treatment with an insurance approval process for DAA coverage once HCV viremia was documented in the recipient. The timing of DAA initiation, duration of recipient viremia, and associated costs incurred by the patient and the institution were assessed in 89 abdominal organ transplant recipients who did not have their DAA treatment subsidized compared to 62 thoracic organ transplant recipients who received DAA treatment that was initially subsidized by the institution. Their analysis showed that by not waiting to initiate DAA treatment for insurance authorization after HCV viremia was documented in the recipient enabled earlier treatment initiation (median, 4 days [IQR, 2-7] vs 10 days [IQR, 8-13]; p <0.001) and shorter duration of viremia (median, 16 days [IQR, 12-29] vs 36 [IQR, 30-47]; p <0.001) (Stewart, 2021).
Selection of the DAA therapy for HCV-negative recipients of a non-liver allograft(s) from an HCV-viremic donor should follow the same principles as for those who develop recurrent HCV infection post liver transplantation (see Patients Who Develop Recurrent HCV Infection Post Liver Transplantation). Importantly, since genotyping of HCV-viremic donors is not routinely performed, only pangenotypic regimens should be utilized if a prophylactic/preemptive treatment approach is used. If treatment is delayed until the recipient has quantifiable HCV RNA, the recipient’s genotype can be used to guide DAA treatment selection if a pangenotypic regimen is not used. Selection of regimens that avoid the use of ribavirin (to reduce ribavirin-associated side effects) and regimens that do not require baseline RAS testing are preferred. Thus, although there are data supporting the safety and efficacy of elbasvir/grazoprevir among HCV-negative kidney and heart transplant recipients of allografts from HCV-viremic donors, the regimen is designated an alternative regimen due to the necessity for baseline RAS testing and its limited genotype coverage. Similarly, ledipasvir/sofosbuvir is designated as an alternative regimen due to lack of pangenotypic coverage.
Notably, organs from HCV-viremic donors may be used in transplant candidates with current or prior HCV infection (see Patients Who Develop Recurrent HCV Infection Post Liver Transplantation).
The interactions of DAA agents and calcineurin inhibitors are complex and unpredictable without formal studies of drug-drug interactions. A summary of interactions between calcineurin inhibitors and DAAs with recommended dosing is provided in the DAA Interactions With Calcineurin Inhibitors table.
Based on the metabolism of grazoprevir and elbasvir, a 15-fold increase in grazoprevir AUC and a 2-fold increase in elbasvir AUC can be expected with cyclosporine coadministration. Therefore, this combination should be avoided. Since a 40% to 50% increase in tacrolimus level is predicted with coadministration of grazoprevir, no dosing adjustments are anticipated but tacrolimus levels should be monitored. No clinically significant drug-drug interactions have been observed between sofosbuvir-inclusive regimens and tacrolimus.
Chronic hepatitis C is independently associated with the development of chronic kidney disease (CKD) (Rogal, 2016); (Fabrizi, 2015). A meta-analysis demonstrated that chronic HCV infection was associated with a 51% increase in the risk of proteinuria and a 43% increase in the incidence of CKD (Fabrizi, 2015). There is also a higher risk of progression to end-stage renal disease (ESRD) in persons with chronic HCV infection and CKD, and an increased risk of all-cause mortality in persons on dialysis (Lee, 2014); (Fabrizi, 2012).
Successful HCV antiviral treatment improves clinical outcomes. Antiviral therapy was associated with a survival benefit among persons on dialysis in a nationwide Swedish registry study (Söderholm, 2018). In a retrospective cohort analysis utilizing the Truven Health MarketScan Database (2008-2015), HCV treatment was associated with a 30% decreased risk of developing CKD (HR, 0.70; 95% CI, 0.55-0.88). Persons with HCV infection experienced a twofold and a 17-fold higher risk of membranoproliferative glomerulonephritis (HR, 2.23; 95% CI, 1.84-2.71) and cryoglobulinemia (HR, 16.91; 95% CI, 12.00-23.81), compared with persons without HCV (Park, 2018).
Among diabetic patients with ESRD receiving care at 4 US health systems, achieving a sustained virologic response (SVR) reduced the risk of developing extrahepatic manifestations of HCV disease, regardless of cirrhosis (sHR=0.46), compared to untreated patients (Li, 2019). In a retrospective observational cohort study, predictors of eGFR improvement after antiviral therapy included baseline CKD (eGFR <60 mL/min) and not having diabetes (Sise, 2019). A prospective cohort study that evaluated estimated glomerular filtration rate in patients with eGFR >15 mL/min demonstrated a lower risk of ESRD in patients who achieve SVR12 (Liu, 2022).
Recommendation for Patients With CKD Stagea |
|
|---|---|
| RECOMMENDED |
RATING |
| No dose adjustment in direct-acting antivirals is required when using recommended regimens.b | I, A or IIa, Bc |
|
a Chronic kidney disease (CKD) stages: 1 = normal (eGFR >90 mL/min); 2 = mild CKD (eGFR 60-89 mL/min); 3 = moderate CKD (eGFR 30-59 mL/min); 4 = severe CKD (eGFR 15-29 mL/min); 5 = end-stage CKD (eGFR <15 mL/min) b A ribavirin dose reduction may be required for patients with CKD stage 3, 4, or 5; see prescribing information for details. c The rating is I, A for patients with CKD stage 1, 2, or 3 and IIa, B for those with CKD stage 4 or 5. |
|
The EXPEDITION-4 trial evaluated the safety and efficacy of 12 weeks of the pangenotypic NS3/NS4A protease inhibitor glecaprevir and the pangenotypic NS5A inhibitor pibrentasvir for genotype 1, 2, 3, 4, 5, or 6 infection (Gane, 2017b). This open-label study enrolled treatment-naive and -experienced patients (previous interferon or peginterferon ± ribavirin, or sofosbuvir and ribavirin ± peginterferon) with CKD stage 4/5, including those with hemodialysis dependence. Baseline characteristics of the 104 patients enrolled in the study were 76% male; 25% black; 19% compensated cirrhosis; 40% treatment experienced; and 82% hemodialysis dependent. The genotype distribution was 22% genotype 1a; 28% genotype 1b; 16% genotype 2; 11% genotype 3; 19% genotype 4; 1% genotype 5; and 1% genotype 6. The daily fixed-dose combination of glecaprevir (300 mg)/pibrentasvir (120mg) was administered as three 100 mg/40 mg fixed-dose combination pills.
The study reported ITT and mITT SVR12 rates of 98% and 100%, respectively. There were no virologic failures. Two patients did not achieve SVR12; 1 patient discontinued the study due to diarrhea in the context of recent gastrointestinal bleeding and the other experienced a cerebral hemorrhage due to uncontrolled hypertension (had achieved SVR4). Adverse events included pruritus (20%), fatigue (14%), and nausea (12%). There were no serious adverse events related to the study drugs, and there were no grade 4 laboratory abnormalities reported. The EXPEDITION-4 trial supports the efficacy and safety of glecaprevir/pibrentasvir in patients with CKD, including ESRD. The recommended duration of therapy is the same as for patients without CKD.
EXPEDITION-5 evaluated the efficacy and safety of fixed-dose glecaprevir/pibrentasvir for chronic HCV infection in adults without cirrhosis or with compensated cirrhosis and stage 3b, 4, or 5 CKD. Among the 101 study participants, 76% (n=77) were on dialysis and 24% (n=24) had predialysis CKD. Fifty-five percent of patients had genotype 1, 27% had genotype 2, 15% had genotype 3, and 4% had genotype 4; no patients had genotype 5 or 6 infection. Eighty-four patients were treated for 8 weeks, 13 patients for 12 weeks, and 4 patients for 16 weeks. The overall SVR12 was 97% (98/101) with no reported virologic failures (Lawitz, 2020).
An integrated analysis of the efficacy and safety of glecaprevir/pibrentasvir in persons with genotypes 1 through 6 and CKD stage 3b, 4, or 5 was performed based on the EXPEDITION-4 and EXPEDITION-5 clinical trials. This analysis included 205 patients with compensated liver disease (with and without cirrhosis) and an eGFR <30 mL/min (EXPEDITION-4) or <45 mL/min (EXPEDITION-5). The majority of patients were treatment naive (69%), with genotype 1 (54%), and on dialysis (79%). In this integrated analysis, 100% SVR12 (mITT) was found with glecaprevir/pibrentasvir therapy in patients with chronic hepatitis C and severe renal impairment regardless of treatment duration (Lawitz, 2018).
Colchicine‐induced rhabdomyolysis due to interaction with glecaprevir/pibrentasvir has been reported in while receiving treatment of gout. Despite a 50% dose reduction of colchicine before initiation of HCV therapy, the patient experienced rhabdomyolysis. This potential interaction with colchicine has the potential for increased risk for muscle toxicity and should prompt consideration of discontinuation of colchicine during therapy, especially in patients with renal insufficiency (Harrison, 2020).
In November 2019, the US FDA amended the package inserts for sofosbuvir-containing regimens to allow use in patients with renal disease, including those with an eGFR ≤30 mL/min and those on dialysis.
A retrospective evaluation of clinical trial participants in 76 clinical trials treated with sofosbuvir with an estimated glomerular filtration rate (eGFR) of 30-89 mL/min/1.73 m2 in nationally-representative administrative claims database demonstrated that participants with CKD did not experience worsening eGFR during sofosbuvir-based treatment, and sofosbuvir was not associated with an increased risk of ESRD in patients with CKD (Sulkowski, 2022). In a Taiwan real-world HCV registry program of 12,995 persons with a prospective evaluation of serial eGFR levels during and following treatment, sofosbuvir was not associated with eGFR change (Huang, 2022).
A prospective multicenter, open-label evaluation of ledipasvir/sofosbuvir (90mg/400mg) daily in patients with HCV with end-stage kidney disease on dialysis demonstrated safety and effectiveness at 8 wks (genotype 1 naïve without cirrhosis), 12 weeks (treatment-experienced genotype 1 treatment-naive or experienced nongenotype 1 without cirrhosis) and 24 weeks (genotypes 1, 2, 4 with compensated cirrhosis). Ninety-four percent (89/95) achieved sustained virologic response 12 weeks after treatment. Six patients died during treatment, however no deaths were related to treatment (Huang, 2022).
A real-world case series of treatment-naive and -experienced patients demonstrated that 12 weeks of sofosbuvir/velpatasvir administered in persons with any genotype and on dialysis resulted in 95% (56/59) SVR12. There were no treatment-related discontinuations or serious adverse events. There were 2 virologic relapses; 1 was associated with nonadherence (Borgia, 2019). A retrospective analysis of 31 treatment-naive patients on hemodialysis demonstrated that 12 weeks of sofosbuvir/velpatasvir administered in persons with any genotype (68% with genotype 1) resulted in a 95% (30/31) SVR12. There was a single virologic relapse among the 3 persons with cirrhosis (Gaur, 2020). A systematic review and meta-analysis of 717 patients with CKD stage 4/5 (58.4% on dialysis) treated with sofosbuvir regimens across 21 studies demonstrated a pooled SVR 12/24 of 97% and a serious adverse event rate of 4.8%. Cirrhotic and noncirrhotic patients achieved comparable SVR rates (Li, 2019a).
Rare adverse advents have been reported among patients with CKD receiving DAAs. Colchicine-induced rhabdomyolysis has been reported in a patient with renal dysfunction being treated with ledipasvir/sofosbuvir while continuing atorvastatin (Patel, 2016). Acute interstitial nephritis following DAA treatment has been described in association with sofosbuvir/ledipasvir (n=5), elbasvir/grazoprevir (n=2), and sofosbuvir/simeprevir (n=1) (Duque, 2021).
The C-SURFER trial evaluated the safety and efficacy of 12 weeks of the daily fixed-dose combination of elbasvir (50 mg)/grazoprevir (100 mg) versus placebo among genotype 1 patients with CKD stage 4 or 5 (eGFR <30 mL/min). The initial study randomized eligible patients to immediate or deferred treatment with elbasvir/grazoprevir. The delayed treatment arm initially received placebo and was later treated with elbasvir/grazoprevir. Elbasvir and grazoprevir are primarily metabolized in the liver and undergo minimal renal elimination.
The data for the immediate treatment arm have been published (Roth, 2015). Seventy-five percent of the study participants were on hemodialysis, and 45% were African American. A small number of patients with compensated cirrhosis were included. Intention-to-treat (ITT) and modified intention-to-treat (mITT) SVR12 rates were 94% and 99%, respectively. There were no changes in erythropoietin use, hemoglobin, or other adverse events in the treatment groups compared to placebo. None of the genotype 1a patients with baseline NS5A resistance-associated substitutions (RASs) experienced viral relapse. The only reported relapse occurred in a patient with genotype 1b. The basis for the lack of impact of NS5A RASs on SVR rates in this population is unclear but may relate to the moderately increased area under the curve (AUC) with grazoprevir and elbasvir observed in patients with stage 4/5 CKD (Zepatier prescribing information, 2019). Among 99 patients assigned to deferred treatment 97 (98%) achieved SVR (Bruchfeld, 2017). In patients with genotype 1a, SVR12 was 85% (11/13) among patients with detectable baseline NS5A RASs and 100% (98/98) among those patients without RASs. One serious adverse event occurred during the deferred treatment (interstitial nephritis) that was considered study drug related. Overall, the efficacy of this regimen among patients assigned to deferred treatment reflected the findings of the immediate treatment group, and the overall efficacy remained high in all subgroups including cirrhosis, diabetes, and hemodialysis. These data support no modification of elbasvir plus grazoprevir dosing for patients on hemodialysis. Of the 3 patients who relapsed in both the immediate and deferred treatment groups, 2 had genotype 1a infection with baseline NS5AA RASs, underscoring the importance of baseline NS5A RASs affecting treatment outcome with this regimen (Bruchfeld, 2017).
Based on these data, daily fixed-dose elbasvir/grazoprevir is recommended for the treatment of genotype 1 in patients with severely compromised renal function. While C-SURFER did not evaluate patients with genotype 4, it is likely that the high efficacy of elbasvir/grazoprevir in genotype 1 and 4 infection in persons with normal renal function can be extrapolated to persons with genotype 4 and CKD stage 4/5. Treatment with elbasvir/grazoprevir in persons with CKD has been shown to be cost-effective in the United States (Elbasha, 2016).
Several real-world studies demonstrated the effectiveness of elbasvir/grazoprevir in persons with genotype 1 or 4 infection. In a retrospective cohort analysis from the TRIO network, 99% (113/114) of patients with stage 4/5 CKD achieved SVR12 (Flamm, 2018). A nationwide retrospective observational cohort study of patients in the US Veterans Health Administration system identified 5961 patients (42.5% genotype 1a, 55.0% genotype 1b) who completed elbasvir/grazoprevir therapy, including 860 patients with stage 3 CKD, 740 patients with stage 4/5 CKD, and 4361 controls (eGFR ≥60 mL/min). The SVR rates were 97% overall, 96% for those with an eGFR ≥60 mL/min, 98% for patients with stage 3 CKD, and 97% for participants with stage 4/5 CKD. No statistically significant differences were found in the SVR rates in persons with or without dialysis among the stage 4/5 CKD patients (adjusted OR 0.91; 95% CI 0.56-1.47 and OR 1.74; 95% CI 0.63-4.81) compared with those with an eGFR ≥60 mL/min (Choi, 2020).
Elbasvir, grazoprevir, and ledipasvir are primarily metabolized in the liver and undergo minimal renal elimination. While exposures to many of these agents are higher in severe renal impairment—presumably due to effects of uremic toxins, parathyroid hormone, and/or cytokines on hepatic metabolism—dose adjustments are not required in the setting of renal impairment.
Recommended and alternative regimens listed by pangenotypic, evidence level and alphabetically for:
Treatment-Naive and Non-DAA-Experienced Kidney Transplant Patients With Genotype 1-6 Infection, With or Without Compensated Cirrhosisa
|
||
|---|---|---|
| RECOMMENDED | DURATION |
RATING |
| Daily fixed-dose combination of glecaprevir (300 mg)/pibrentasvir (120 mg)b | 12 weeks |
I, Ac IIa, Cd |
| Daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg) | 12 weeks | IIa, C |
| Genotype 1, 4, 5, or 6 only: Daily fixed-dose combination of ledipasvir (90 mg)/sofosbuvir (400 mg) | 12 weeks | I, A |
| ALTERNATIVE | DURATION |
RATING |
| Genotype 1 or 4 only: Daily fixed-dose combination of elbasvir (50 mg)/ grazoprevir (100 mg) for patients without baseline NS5A RASse for elbasvir | 12 weeks | I, B |
|
a For decompensated cirrhosis, please refer to the appropriate section. b Dosing is 3 coformulated tablets (glecaprevir [100 mg]/pibrentasvir [40 mg]) taken once daily. Please refer to the prescribing information. c Based on evidence for patients without cirrhosis. d Based on evidence for patients with compensated cirrhosis. e Includes genotype 1a resistance-associated substitutions at amino acid positions 28, 30, 31, or 93 known to confer antiviral resistance. |
||
Recommended regimen for:DAA-Experienced Kidney Transplant Patients With Genotype 1-6 Infection, With or Without Compensated Cirrhosisa |
||
|---|---|---|
| RECOMMENDED | DURATION |
RATING |
|
Daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg)/voxilaprevir (100 mg), with or without ribavirinb |
12 weeks | IIa, C |
|
a Excludes CTP class B and class C patients. For decompensated cirrhosis, please refer to the appropriate section. b For patients with cirrhosis and multiple negative baseline characteristic, consideration should be given to adding ribavirin. If renal dysfunction is present, a lower starting dose is recommended. Maximum ribavirin dose is 1000 mg/d for patients who weigh <75 kg and 1200 mg/d for those who weigh ≥75 kg. |
||
For additional information on treatment of DAA failures post transplant, treatment of decompensated cirrhosis following transplantation, treatment of transplant recipients from HCV-positive donors, and post-transplant drug-drug interactions, please see Patients Who Develop Recurrent HCV Infection Post Liver Transplantation.
The phase 3, open-label, single arm MAGELLAN-2 study evaluated a 12-week course of the pangenotypic regimen of glecaprevir/pibrentasvir in 100 liver (n=80) and kidney (n=20) transplant recipients with genotypes 1-6 infection who were at least 3 months post transplant. Cirrhotic patients were excluded. SVR12 was achieved in 98% of patients; a single patient experienced virologic failure (Reau 2018). The safety profile was excellent with 1 treatment discontinuation for an adverse event not considered to be therapy related. One rejection episode occurred in a liver transplant recipient. While glecaprevir/pibrentasvir is an effective pangenotypic regimen as demonstrated in the nontransplant population, there were no genotype 5 transplant recipients in the study.
There are potential drug-drug interactions with cyclosporine. Review the DAA interactions with calcineurin inhibitors table in the post liver transplantation section before prescribing HCV DAA therapy to a renal transplant recipient.
A recent phase 2, open-label clinical trial evaluated the safety and efficacy of the daily fixed-dose combination of ledipasvir (90 mg)/sofosbuvir (400 mg) in 114 kidney transplant recipients who were more than 6 months post transplant (Colombo, 2017). Enrolled patients had genotype 1 (91%) or 4 infection; 69% were treatment naive and 15% had compensated cirrhosis. Patients were randomized to 12 weeks or 24 weeks of ledipasvir/sofosbuvir. Median eGFR prior to treatment was 50 mL/min for patients in the 12-week study arm and 60 mL/min for those in the 24-week arm. Overall SVR12 was 100% (114/114). Adverse events were common (64%) and serious adverse events occurred in 13 patients (11%); a single participant discontinued treatment because of an adverse event. Four patients with an eGFR >40 mL/min at baseline experienced a decrease to <30 mL/min during therapy. The eGFR increased to >30 mL/min at the last visit recorded in 3 of these patients; 1 patient who had interrupted study treatment had a final eGFR of 14.4 mL/min. All but 1 of the 6 patients with compensated cirrhosis whose eGFR decreased to <40 mL/min continued study treatment without interruption; none permanently discontinued study treatment.
Several additional reports have described successful outcomes with combination direct-acting antiviral (DAA) therapy in kidney transplant recipients (Saxena, 2017); (Sawinski, 2016). One study evaluated treatment safety and efficacy among 20 HCV-infected kidney transplant recipients (88% genotype 1; 50% with advanced fibrosis; 60% treatment-experienced with an interferon-based regimen) who received sofosbuvir-based therapy. Various regimens were used, including simeprevir plus sofosbuvir (n=9); ledipasvir/sofosbuvir (n=7); sofosbuvir plus ribavirin (n=3); and daclatasvir plus sofosbuvir (n=1). SVR12 was 100% (Sawinski, 2016). Two patients required dose reductions due to anemia associated with ribavirin use. However, no significant changes in serum creatinine or proteinuria, or graft rejection were seen before or after treatment. Forty-five percent of patients required dose reduction of immunosuppressive agents while on antiviral therapy.
Real-world data from the ongoing HCV-TARGET study have also demonstrated the efficacy of DAA therapy in patients with kidney transplant and in those with dual liver and kidney transplant (Saxena, 2017). Various regimens were used, including sofosbuvir/ledipasvir ± ribavirin (85%); sofosbuvir plus daclatasvir ± ribavirin (9%); and ombitasvir/paritaprevir/ritonavir plus dasabuvir ± ribavirin (6%). SVR12 was 95% in those with kidney transplant and 91% in dual liver and kidney transplant recipients.
No change in calcineurin inhibitor dose is needed for patients receiving ledipasvir/sofosbuvir. Review the DAA interactions with calcineurin inhibitors table in the post liver transplantation section before prescribing HCV DAA therapy to a renal transplant recipient.
There are no published clinical trials regarding the use of the fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg) in kidney transplant recipients. There are, however, significant data addressing the efficacy and safety of this regimen in the nontransplant and liver transplant settings.
In liver transplant recipients (discussed in Patients who Develop Recurrent HCV Infection Post Liver Transplantation), the safety and efficacy of sofosbuvir/velpatasvir for 12 weeks was evaluated in 79 patients (n=5 with cirrhosis; n=4 DAA experienced) with genotype 1-4 infection (Agarwal, 2018). Treatment was well-tolerated with 99% of patients completing treatment. SVR12 rates by genotype were 93% genotype 1a (n=15); 96% genotype 1b (n=22); 100% genotype 2 (n=3); 97% genotype 3 (n=35); and 100% genotype 4 (n=4).
In the nontransplant setting (discussed in detail in the Initial and Retreatment sections), the phase 3, double-blind, placebo-controlled ASTRAL-1 study demonstrated an overall SVR of 99% among 742 treatment-naive or -experienced patients with genotype 1, 2, 4, 5, or 6 infection (Feld, 2015). In the phase 3, open-label ASTRAL-3 study, 552 treatment-naive or -experienced patients with genotype 3 (with or without compensated cirrhosis) were randomized in a 1:1 ratio to 12 weeks of sofosbuvir/velpatasvir or 24 weeks of sofosbuvir plus weight-based ribavirin. SVR12 was 95% for the sofosbuvir/velpatasvir treatment arm, which was superior to the SVR12 80% among patients receiving sofosbuvir plus ribavirin for 24 weeks (Foster, 2015a).
No change in calcineurin inhibitor dose is needed for patients receiving sofosbuvir/velpatasvir. Review the DAA interactions with calcineurin inhibitors table in the post liver transplantation section before prescribing HCV DAA therapy to a renal transplant recipient.
To date, there are no published clinical trials evaluating use of the daily fixed-dose combination of sofosbuvir (400 mg)/velpatasvir (100 mg)/voxilaprevir (100 mg) in kidney transplant recipients. There are, however, significant data addressing the efficacy and safety of this regimen in the nontransplant setting (Degasperi, 2019); (Llaneras, 2019); (Bourliere, 2017); (Jacobson, 2017); (Soriano, 2017); (Saxena, 2016).
Two phase 3, open label, randomized clinical trials were conducted to determine the safety and efficacy of sofosbuvir/velpatasvir/voxilaprevir in nontransplant patients previously treated with a DAA regimen. The POLARIS-1 study included nontransplant patients who had previously received a regimen containing and NS5A inhibitor. Patients were randomized to 12 weeks of sofosbuvir/velpatasvir/voxilaprevir or placebo. SVR for patients on active treatment was 96%. POLARIS-4 compared 12 weeks of sofosbuvir/velpatasvir/voxilaprevir to 12 weeks of sofosbuvir/velpatasvir in non-NS5A inhibitor DAA-experienced nontransplant patients (Bourliere, 2017). Overall, 69% of participants were previously exposed to sofosbuvir plus ribavirin ± peginterferon, and 11% were exposed to sofosbuvir plus simeprevir. Cirrhosis was common, 46% in both study arms. SVR12 rates were 98% with sofosbuvir/velpatasvir/voxilaprevir and 90% with sofosbuvir/velpatasvir.
Velpatasvir is a substrate for CYP3A4, CYP2C8, and CYP2B6, a weak inhibitor of P-gp and OATP transporters, and a moderate inhibitor of the breast cancer resistance protein (BCRP) membrane transporter. As such, velpatasvir is moderately affected by potent inhibitors and, to a greater extent, potent inducers of enzyme/drug transporter systems (Mogalian, 2016). Based on this profile, which is similar to ledipasvir, clinically significant drug-drug interactions would not be expected for coadministration of sofosbuvir/velpatasvir with common immunosuppressive agents (eg, tacrolimus, cyclosporine, corticosteroids, mycophenolate mofetil, or everolimus). Review the DAA interactions with calcineurin inhibitors table in the post liver transplantation section before prescribing HCV DAA therapy to a renal transplant recipient.
Data from small, real-world studies evaluating elbasvir/grazoprevir are available. One such study evaluated 11 kidney transplant recipients with significant kidney function impairment (GFR <40 mL/min) treated with elbasvir/grazoprevir for 12 to 16 weeks. SVR12 was 100% (Eisenberger, 2019).
There are significant drug-drug interactions with cyclosporine. Review the DAA interactions with calcineurin inhibitors table in the post liver transplantation section before prescribing HCV DAA therapy to a renal transplant recipient.
Acute hepatitis C infection is most often asymptomatic and frequently develops into chronic infection. Case reports of acute hepatitis C have increased in the US since 2010 and have most often been associated with parenteral exposures to blood or body fluids (CDC, 2019). Although HCV infection is primarily associated with injection drug use, certain behaviors (eg, unprotected [without a condom] receptive anal intercourse)—primarily among men who have sex with men—are risk factors for transmission (Lockart, 2019); (Price, 2019). The syndemic of opioid use disorder and HCV and HIV transmission contributes to the burden of disease in certain populations (Butt, 2020).
Recommended Testing for Diagnosing Acute HCV Infection |
|
|---|---|
| RECOMMENDED |
RATING |
| HCV antibody and HCV RNA testing are recommended when acute HCV infection is suspected due to exposure, clinical presentation, or elevated aminotransferase levels (see Testing Algorithm figure). | I, C |
Recommendations for HCV testing are also found in the Testing and Linkage to Care section.
Diagnosis of acute HCV infection enables estimation of annual incidence rates and transmission patterns, thereby facilitating implementation and assessment of prevention programs. At the individual level, a diagnosis of acute infection expedites linkage to care, counseling regarding high-risk behavior, and timely interventions to reduce virus transmission and liver disease progression (Bruneau, 2014). Some persons involved in high-risk behaviors practice serosorting, defined as using HCV antibody serostatus to determine whether to engage in high-risk behaviors with certain individuals (Smith, 2013). Thus, undiagnosed acutely infected persons may be at greater risk of transmitting HCV to their presumably seronegative contacts than would be expected by chance.
The best laboratory evidence to support a diagnosis of acute HCV infection is a positive HCV RNA test in the setting of a negative HCV antibody test (identification during the seronegative window period) (Cox, 2005), or a positive HCV antibody test after a prior negative HCV antibody test (seroconversion). There are rare instances in which these approaches may be misleading, such as in immunosuppressed individuals with impaired antibody production (Chamot, 1990).
Discrete Exposure
The aforementioned types of clear, laboratory-based documentation of acute HCV infection are most easily achieved when there has been a discrete, known or suspected exposure (eg, after new onset or a change in drug injection practice, a percutaneous needle-stick exposure to an HCV-infected individual, a potentially nonsterile tattoo, or sexual assault). In those instances, baseline HCV antibody and RNA testing should be done within 48 hours of the exposure to document whether there was antecedent HCV infection (see Testing Algorithm figure).
If baseline testing is negative, repeat testing is recommended. Frequency of testing can be tailored based on management objectives (eg, monthly testing to identify and treat acute infection). If baseline HCV antibody testing is positive but RNA testing is negative, repeat HCV RNA and alanine aminotransferase (ALT) testing is recommended to identify an acute reinfection. When baseline HCV antibody and RNA testing are both positive, the person most likely already has chronic HCV infection from prior exposure(s).
No Discrete Exposure
Individuals suspected of having acute HCV infection often do not have a discrete exposure or have no prior baseline testing, making a diagnosis of acute infection more difficult (see Blood Test Interpretation Table). Acute infection should be suspected if there is a new rise in the ALT level without an alternate cause (Blackard, 2008); (Kim, 2013). Acute infection should also be suspected when there are low (especially <104 IU/mL) or fluctuating (>1 log10 IU/mL) HCV RNA values, or spontaneous clearance. These patterns do not commonly occur outside of the first 6 months after HCV infection (McGovern, 2009). In those with a high index of suspicion for HCV exposure (eg, recently relapsed injection drug use, other high-risk exposure), an HCV PCR should be repeated, if negative.
Patients suspected of having acute HCV infection should also have a laboratory evaluation to exclude other or coexisting causes of acute hepatitis (eg, hepatitis A virus, hepatitis B virus, hepatitis delta virus if chronically infected with hepatitis B, and autoimmune hepatitis) (Kushner, 2015). In patients with sexual acquisition of acute HCV, evaluation for concurrent genital ulcerative disease and proctitis is recommended (Todesco, 2019); (Goldenberg, 2017). Patients should also have HIV testing.
Table. Interpretation of Blood Tests for Diagnosis of Acute HCV Infection
| TEST | INTERPRETATION FOR DIAGNOSIS OF ACUTE HCV |
|---|---|
| HCV Antibody |
|
| HCV RNA |
|
| ALT |
|
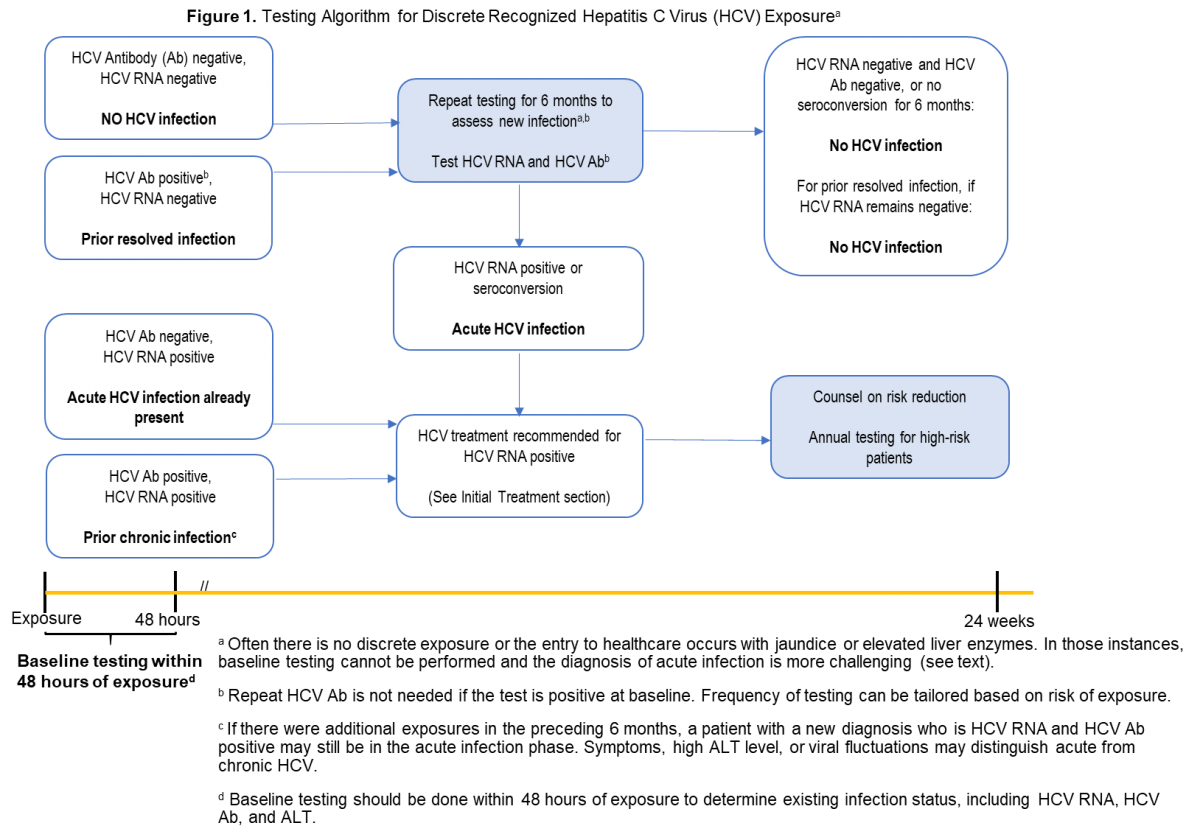
Pharmacologic Prophylaxis Not Recommended |
|
|---|---|
| NOT RECOMMENDED |
RATING |
| Pre-exposure or post-exposure prophylaxis with antiviral therapy is not recommended. | III, C |
There are no data on the efficacy or cost-effectiveness of antiviral therapy for pre-exposure or post-exposure prophylaxis of HCV infection.
Recommendations for Medical Management and Monitoring of Acute HCV Infection |
|
|---|---|
| RECOMMENDED |
RATING |
| After the initial diagnosis of acute HCV with viremia (defined as quantifiable RNA), HCV treatment should be initiated without awaiting spontaneous resolution. | I, B |
| Counseling is recommended for patients with acute HCV infection to avoid hepatotoxic insults, including hepatotoxic drugs (eg, acetaminophen) and alcohol consumption, and to reduce the risk of HCV transmission to others. | I, C |
| Referral to an addiction medicine specialist is recommended for patients with acute HCV infection related to substance use. | I, B |
Patients with acute HCV infection should be treated upon initial diagnosis without awaiting spontaneous resolution, using a “test and treat” strategy and according to the simplified approach, if eligible. Real-world data have demonstrated a reduction in HCV viremia prevalence and incidence with unrestricted access to HCV therapy (Boerekamps, 2018). In addition, mathematical modeling suggests that DAA treatment scale-up, especially among those at highest risk of transmission, can reduce HCV incidence and prevalence (Martin, 2013); (Martin, 2016). Moreover, delay introduced by waiting for spontaneous clearance may be associated with loss to follow up.
Individuals with acute HCV should be counseled to reduce behaviors that could result in virus transmission, such as sharing injection equipment and engaging in high-risk sexual practices. Because the risk of transmission of other bloodborne, sexually transmitted infections (eg, HIV and HBV) is higher in the acute infection phase, some experts counsel patients with acute HCV to consider using barrier precautions, even in a stable monogamous relationship (see Testing and Linkage to Care). For individuals with acute HCV infection who have a history of recent injection drug use, referral to harm reduction services and an addiction medicine specialist is recommended when appropriate (Litwin, 2009); (Strathdee, 2005).
Patients with acute hepatitis C are often asymptomatic or have nonspecific symptoms (eg, fatigue, anorexia, mild or moderate abdominal pain, low-grade fever, nausea, and/or vomiting) that frequently are not recognized as being associated with acute HCV infection. A small proportion (<25%) of patients with acute HCV develop jaundice. Patients diagnosed with acute HCV should initially be monitored with hepatic panels (ALT, aspartate aminotransferase [AST], bilirubin, and international normalized ratio [INR] in the setting of an increasing bilirubin level) at 2- to 4-week intervals (Blackard, 2008). With treatment, a rapid improvement of laboratory parameters is expected.
There is no need to alter concomitant medications that are metabolized by hepatic enzymes unless there is concern for developing acute liver failure (eg, increasing bilirubin level and INR). Acetaminophen and alcohol consumption should be avoided during acute HCV infection (Proeschold-Bell, 2012); (Dieperink, 2010); (Whitlock, 2004).
Hospitalization is rarely indicated unless nausea and vomiting are severe. Although acute liver failure is very rare (<1%), it represents a serious and life-threatening complication of acute HCV infection. Patients with an INR >1.5 and those who exhibit any signs of acute liver failure (eg, hepatic encephalopathy) should be referred to a liver transplant center immediately. Use of HCV antiviral regimens in acute liver failure should be managed by a clinician experienced in HCV treatment, ideally in consultation with a liver transplant specialist.
HCV infection spontaneously clears in 20% to 50% of patients (Kamal, 2008). Clearance of acute HCV infection occurs within 6 months of the estimated time of infection (median, 16.5 weeks) in at least 2/3 of patients who spontaneously clear HCV. Only 11% of those who remain viremic at 6 months will spontaneously clear the infection at a later time (Grebely, 2014). Patients who have spontaneously cleared should not be treated with antiviral therapy. However, they should be counseled about the possibility of reinfection and tested routinely for this development if risk behaviors are ongoing (see Testing and Linkage to Care). Of note, transient suppression of viremia can occur in those with acute HCV infection, even among those who progress to chronic infection. Thus, a single undetectable HCV RNA test result is insufficient to declare spontaneous clearance (see Testing and Linkage to Care) (Villano, 1999); (Mosley, 2008).
Predictors of spontaneous clearance include jaundice, elevated ALT level, hepatitis B virus surface antigen (HBsAg) positivity, female sex, younger age, genotype 1 infection, and host genetic polymorphisms, most notably those near the IL28B gene (Kamal, 2008); (Mosley, 2008).
Recommended Regimens for Patients With Acute HCV Infection |
|
|---|---|
| RECOMMENDED |
RATING |
| Owing to high efficacy and safety, the same regimens that are recommended for chronic HCV infection are recommended for acute infection. | IIa, C |
A number of studies have evaluated DAA treatment of acute HCV infection. Small single-arm, uncontrolled studies have evaluated 6 or 8 weeks of ledipasvir/sofosbuvir. One such study demonstrated 100% SVR with 8 weeks of ledipasvir/sofosbuvir among 27 men with acute HCV and HIV-coinfection (Naggie, 2019). Investigators conducting another study evaluated 6 weeks of ledipasvir/sofosbuvir in a similar cohort (25/26 with HIV coinfection). Among participants with genotype 1 infection, 79% (15/19) achieved SVR12; 71% (5/7) of those with genotype 4 infection achieved SVR12 with this shortened regimen. Among the 6 individuals whose treatment did not lead to SVR12, there were 3 relapses (all had baseline HCV RNA levels >7 log10 IU/mL). Three participants achieved SVR4 but were lost to follow-up (Rockstroh, 2017b). A phase 2 study followed a similar treatment protocol (ie, 6 weeks of ledipasvir/sofosbuvir) among 20 individuals with genotype 1 HCV monoinfection, all of whom achieved SVR12 (Deterding, 2017).
An open-label, single-arm, multicenter pilot study evaluated the efficacy of 6 weeks of the pangenotypic regimen glecaprevir/pibrentasvir among persons with acute/recent HCV infection (ie, duration of infection <12 months). SVR12 was 90% (27/30); a single virological failure occurred in a man with genotype 1a, HIV coinfection, and a viral load of 7.7 log10 IU/mL. This patient was successfully retreated (Martinello, 2020).
In the only randomized trial to date, investigators compared 6 vs 12 weeks of sofosbuvir/velpatasvir in the international REACT trial of acute/recent infection. The study was stopped early due to inferiority of the shortened (ie, 6 week) arm. In the 6-week arm, 81.7% (76/93) (on ITT) and 89.4% (76/85) (on mITT) of participants achieved SVR with 6 relapses and 8 nonvirologic failures. In the 12-week group, 90.5% (86/95) on ITT and 97.7% (86/88) (on mITT) achieved SVR with no virologic failures (3 participants were lost to follow-up). There were no clear predictors of relapse aside from shorter treatment duration (Matthews, 2021).
To date, there are insufficient data to support a particular regimen or treatment duration outside of a clinical trial. Until more definitive data are available, treatment as described for chronic hepatitis C is recommended (see Initial Treatment of HCV Infection). Pangenotypic regimens are recommended if HCV genotyping is unavailable or if concern of exposure to more than 1 genotype exists. Using the same regimens to treat acute/recent HCV as for chronic HCV infection also simplifies management, as defining acute HCV may be clinically challenging.
Recommendation for Universal Hepatitis C Screening in Pregnancy |
|
|---|---|
| RECOMMENDED |
RATING |
| As part of prenatal care, all pregnant persons should be tested for HCV infection with each pregnancy, ideally at the initial visit. (See Recommendations for Initial HCV Testing and Follow-Up.) | I, B |
It has been estimated that up to 29,000 HCV-infected women gave birth each year from 2011 to 2014 (Ly, 2017). Additionally, there has been an increase in HCV among young adults, including women of childbearing age (Watts, 2017); (Koneru, 2016); (Kuncio, 2016). Identifying HCV infection as women engage in prenatal care would allow for appropriate assessment of liver disease status and ideally facilitate linkage to HCV care after delivery. In addition, prenatal HCV diagnosis is a prerequisite for appropriate screening and care for exposed children. Risk factor-based HCV screening has never been shown to be effective (Kuncio, 2015); (Waruingi, 2015); (Fernandez, 2016) and inconsistent screening and counseling practices have been reported among obstetricians and gynecologists (Boaz, 2003). Consequently, the US Preventative Task Force (Owens, 2020), US Centers for Disease Control (Schillie, 2020), have published recommendations for universal HCV screening of all adults, including screening during prenatal care. Recently, the American College of Obstetricians and Gynecologists have issued the Practice Advisory to test all women at the beginning of each pregnancy for HCV (https://www.acog.org/clinical/clinical-guidance/practice-advisory/articles/2021/05/routine-hepatitis-c-virus-screening-in-pregnant-individuals). Testing at the initiation of prenatal care is considered optimal to maximize opportunities for education, referral, and appropriate testing for the exposed infant. Early identification is key as women living with HCV and their exposed infants are at significant risk for not linking to appropriate HCV evaluation or care. Women should be tested with an HCV-antibody test. If positive, this should be followed with testing for HCV RNA.
HCV-infected pregnant persons should be linked to care so that antiviral treatment can be initiated at the appropriate time (see Testing and Linkage to Care section). Recent modeling studies demonstrate that universal HCV screening in pregnancy is cost-effective and would reduce long-term morbidity with linkage to care and treatment (Tasillo, 2019). Infants of HCV-infected women should be tested and followed as described in the HCV in Children section.
The Society for Maternal-Fetal Medicine recommends several obstetrical practices in women with HCV infection, including preference for amniocentesis over chorionic villus sampling when invasive prenatal diagnostic testing is indicated, as well as avoidance of internal fetal monitoring during labor, prolonged rupture of membranes, and episiotomies (Hughes, 2017).
Recommendation Regarding HCV Treatment and Pregnancy |
|
|---|---|
| RECOMMENDED |
RATING |
| For women of reproductive age with known HCV infection, antiviral therapy is recommended before considering pregnancy, whenever practical and feasible, to reduce the risk of HCV transmission to future offspring. | I, B |
Women of reproductive age with HCV should be counseled about the benefit of antiviral treatment prior to pregnancy to improve the health of the mother and eliminate the low risk of mother-to-child transmission (MTCT). People who become pregnant while on DAA therapy (with or without ribavirin) should discuss the risks versus benefits of continuing treatment with their physicians. Ribavirin is contraindicated in pregnancy due to its known teratogenicity. In addition, the risk for teratogenicity persists for up to 6 months after ribavirin cessation and applies to women taking ribavirin and female partners of men taking ribavirin. If exposed to ribavirin, they should also have their maternal and fetal outcomes reported to the ribavirin pregnancy registry (also see Recommended Monitoring for Pregnancy-Related Issues Prior to and During Antiviral Therapy That Includes Ribavirin).
There are no large-scale clinical trials evaluating the safety of direct-acting antivirals (DAAs) in pregnancy. A small study evaluating the pharmacokinetics of ledipasvir/sofosbuvir in pregnancy demonstrated 100% SVR12 and no safety concerns. Similarly, an international case series of 15 pregnant persons treated with ledipasvir/sofosbuvir reported 100% SVR12 and no early safety concerns in the women or their infants (Yattoo, 2018); (Chappell, 2020). Currently, there are no available data on the use of pangenotypic regimens during pregnancy.
Despite the lack of a recommendation, treatment can be considered during pregnancy on an individual basis after a patient-physician discussion about the potential risks and benefits.
Recommendations for Monitoring HCV-Infected Women During Pregnancy |
|
|---|---|
| RECOMMENDED |
RATING |
| HCV RNA and routine liver function tests are recommended at initiation of prenatal care for HCV-antibody–positive pregnant persons to assess the risk of mother-to-child transmission (MTCT) and severity of liver disease. | I, B |
| All pregnant persons with HCV infection should receive prenatal and intrapartum care that is appropriate for their individual obstetric risk(s) as there is no currently known intervention to reduce MTCT. | I, B |
| In HCV-infected pregnant persons with pruritus or jaundice, there should be a high index of suspicion for intrahepatic cholestasis of pregnancy (ICP) with subsequent assessment of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and serum bile acids. | I, B |
| HCV-infected persons with cirrhosis should be counseled about the increased risk of adverse maternal and perinatal outcomes. Antenatal and perinatal care should be coordinated with a maternal-fetal medicine (ie, high-risk pregnancy) obstetrician. | I, B |
Pregnancy Impact on HCV Infection
Pregnancy itself does not appear to negatively affect chronic HCV infection. In general, serum ALT levels decrease during the first and third trimesters of pregnancy and increase after delivery. HCV RNA levels rise during the first and third trimesters, reaching a peak during the third trimester, and decrease postpartum (Conte, 2000); (Gervais, 2000). These effects are likely due to the immunosuppressive effects of pregnancy and increased maternal plasma volume. HCV-infected pregnant persons have a higher incidence of intrahepatic cholestasis of pregnancy (ICP) (pooled OR 20.40 [95% CI, 9.39-44.33, I2=55%]) based on a meta-analysis of 3 studies when compared to noninfected pregnant persons (Wijarnpreecha, 2017). ICP is associated with an increased rate of adverse maternal and fetal outcomes; all patients with this syndrome should be immediately referred to a high-risk obstetrical specialist for monitoring and treatment.
HCV Infection Impact on Pregnancy and Perinatal Outcomes
Although some studies show an increased risk of adverse perinatal outcomes (eg, preterm delivery, low birth weight infants, and congenital anomalies) with maternal HCV infection, these risks are confounded by comorbid conditions, such as substance use (Connell, 2011). However, pregnant persons with cirrhosis are at increased risk for poor maternal outcomes (ie, preeclampsia, cesarean section, hemorrhagic complication, and death) and neonatal outcomes (ie, preterm delivery, low birth weight, and neonatal death) (Puljic, 2016); (Tan, 2008). Women with cirrhosis should be counseled about these increased risks and care should be coordinated with specialists in maternal-fetal medicine.
Hepatitis C MTCT occurs at an overall rate of 5% to 15% (Jhaveri, 2015); (Shebl, 2009); (Mast, 2005); (Ceci, 2001), with the number that progress to chronic infection being 3% to 5%. No specific risk factor predicts transmission and no specific intervention (eg, antiviral, mode of delivery, or others) has been demonstrated to reduce HCV transmission—except for suppression of HIV replication in women with HIV/HCV coinfection (Checa Cabot, 2013). Given the potential associated risk of MTCT, it is advisable to avoid invasive procedures (eg, fetal scalp monitors and forceps delivery).
The neuropsychiatric and systemic side effects of interferon-based agents and the pregnancy category X rating of ribavirin made studies involving these drugs to interrupt MTCT untenable for safety reasons. It is important to note that DAAs have not been formally studied as a way to interrupt MTCT. DAAs have not demonstrated significant toxicity in animal studies, and antiviral medication use has become the standard of care for people with HIV and hepatitis B infection. Therefore, it is realistic to think that DAAs could be used in the future to interrupt MTCT. However, with a low transmission rate, improved methods to identify mothers who are likely to transmit are needed to reduce the number needed to treat below 20 to prevent 1 transmission event. DAA therapy is not recommended during pregnancy to reduce MTCT due to the current lack of safety and efficacy data.
Recommendations Regarding Breastfeeding and Postpartum Care for HCV-Infected Women |
|
|---|---|
| RECOMMENDED |
RATING |
| Breastfeeding is not contraindicated in women with HCV infection, except when the mother has cracked, damaged, or bleeding nipples, or in the context of HIV coinfection. | I, B |
| Women with HCV infection should have their HCV RNA reevaluated after delivery to assess for spontaneous clearance. | I, B |
HCV and Breastfeeding
Breastfeeding is not a risk for HCV MTCT (CDC, 1998) with studies showing similar rates of maternal infection in breast-fed and bottle-fed infants (Resti, 1998). However, given the associated risks of HCV transmission with blood exposure and HIV transmission with breastfeeding, it is recommended that HCV-infected women who breastfeed abstain from doing so while their nipples are cracked, damaged, or bleeding, and in the context of HIV/HCV coinfection.
Spontaneous Clearance in the Postpartum Period
HCV RNA levels can fluctuate during pregnancy and the postpartum period. The most frequently observed pattern is a steady rise in HCV RNA levels during pregnancy followed by a slight or significant drop (>3 to 4 log10) in the postpartum period (Lin, 2000). This is most likely due to the release of tolerance in HCV-specific T lymphocyte responses that develop during pregnancy (Honegger, 2013). Spontaneous clearance of HCV can occur in the postpartum period. Previous studies with small numbers of patients demonstrated that up to 10% of postpartum women became HCV RNA undetectable (Honegger, 2013); (Hattori, 2003); (Lin, 2000). A recent study from Egypt demonstrated a 25% rate of spontaneous resolution that was strongly associated with the favorable IL28B allele (Hashem, 2017).
Given these findings, women should have their HCV RNA re-evaluated after delivery. In that time, HCV RNA could become undetectable or rebound to prepregnancy levels. The possibility of spontaneous viral clearance should be considered for any woman who is being assessed for DAA treatment in the postpartum period.
Recommendations for HCV Testing of Perinatally Exposed Children and Siblings of Children With HCV Infection |
|
|---|---|
| RECOMMENDED |
RATING |
| All children born to HCV-infected women should be tested for HCV infection. Testing is recommended using an antibody-based test at or after 18 months of age. | I, A |
| Repetitive HCV RNA testing prior to 18 months of age is not recommended. | III, A |
| Children who are anti-HCV positive after 18 months of age should be tested with an HCV-RNA assay after age 3 to confirm chronic hepatitis C infection. | I, A |
| The siblings of children with vertically-acquired chronic HCV should be tested for HCV infection, if born from the same mother. | I, C |
Although the prevalence of chronic hepatitis C is lower in children than adults, an estimated 3.5 to 5 million children worldwide have chronic HCV infection (Indolphi, 2019); (Gower, 2014). Data from the National Health and Nutrition Examination Survey (NHANES) indicate that 0.2% of 6- to 11-year-olds (31,000 children) and 0.4% of 12- to 19-year-olds (101,000 adolescents) in the US are HCV antibody positive (Alter, 1999).
As birth to a woman with chronic hepatitis C is a known risk for infection, children born to these women should be evaluated and tested for HCV. The rate of mother-to-child transmission (MTCT) of HCV infection is approximately 5%, although rates are higher among women with inadequately controlled HIV coinfection, and women with higher HCV-RNA levels, (>6 log10 IU/mL) (Benova, 2014); (Delotte, 2014); (Cottrell, 2013); (Shebl, 2009). Identifying, following, and treating exposed children is recommended. The preferred assay for evaluation of HCV infection early in life is HCV-RNA testing, as maternal antibodies and consequently anti-HCV assay positivity may persist for 18 months (Aniszewska, 2012); (England, 2005). About 25% to 50% of infected infants spontaneously resolve HCV infection (loss of previously detectable HCV RNA) by 4 years of age (Indolfi, 2019); (Garazzino, 2014); (Farmand, 2012); (Yeung, 2007); (EPHCVN, 2005); (Mast, 2005).
There is considerable debate about the utility of HCV-RNA testing within the first year of life. Proponents argue that use of a highly sensitive RNA assay early in life can increase the rate of infected infants detected, and that a negative result strongly suggests the infant is not infected while a positive result helps identify HCV cases earlier. Proponents also want to seize opportunity to test in a patient group that is often lost to follow-up. Opponents argue that early testing does not change the need for definitive testing at or after 18 months; HCV RNA is more expensive than an antibody-based test; and there is no intervention or treatment that will occur prior to age 3—because of lack of approved drugs for this age group and to allow for possible spontaneous clearance. One large single center study demonstrated that HCV-RNA testing done in exposed infants aged 2 months to 6 months led to reliable positive and negative results that correlated with ultimate testing at 18 months (Honegger, 2018); (Gowda, 2021). Given these results and the value placed on enhancing HCV elimination efforts by reducing missed opportunities for testing, the panel recommends considering HCV RNA testing as early as 2 months of age. There is no value in repeated HCV-RNA testing prior to 18 months of age, but anti-HCV testing should take place at or after 18 months of age.
Recommendations for Counseling Parents Regarding Transmission and Prevention in Children with HCV Infection |
|
|---|---|
| RECOMMENDED |
RATING |
| Parents should be informed that hepatitis C is not transmitted by casual contact and, as such, children with HCV infection do not pose a risk to other children and can participate in school, sports, and athletic activities, and engage in all other regular childhood activities without restrictions. | I, B |
| Parents should be informed that universal precautions should be followed at school and in the home of children with HCV infection. Educate families and children about the risk and routes of HCV transmission, and the techniques for avoiding blood exposure, such as avoiding the sharing of toothbrushes, razors, and nail clippers, and the use of gloves and dilute bleach to clean up blood. | I, B |
HCV-infected children often face discrimination and stigmatization in school and child-care settings that is driven by public misunderstanding regarding hepatitis C transmission. HCV is not transmitted by casual contact in the absence of blood exposure. Families should not be forced to disclose a child’s HCV infection status, and children should not be restricted from any routine childhood activity.
The risk of sexual transmission of hepatitis C is considered very low/rare. Sexual transmission occurs but is generally inefficient except among HIV-infected men who have unprotected sex with men (see HCV Testing and Linkage to Care) (Tieu, 2018); (Vaux, 2019); (Schmidt, 2014). Adolescents with HIV infection and those with multiple sexual partners or sexually transmitted infections (STIs) should be encouraged to use barrier precautions to prevent sexual transmission of HCV and other STIs. Other adolescents with HCV infection should be counseled that the risk of sexual transmission is low but barrier precautions are recommended for other reasons (see Testing and Linkage to Care: Table 2 - Measures to Prevent Transmission of HCV).
Recommendations for Monitoring and Medical Management of Children With HCV Infection |
|
|---|---|
| RECOMMENDED |
RATING |
| Routine liver biochemistries at initial diagnosis and at least annually thereafter are recommended to assess for disease progression. | I, C |
| Appropriate vaccinations are recommended for children with chronic HCV infection who are not immune to hepatitis B virus and/or hepatitis A virus to prevent these infections. | I, C |
| Disease severity assessment via routine laboratory testing and physical examination, as well as use of evolving noninvasive modalities (ie, elastography, imaging, or serum fibrosis markers) is recommended for all children with chronic HCV infection. | I, B |
| Children with cirrhosis should undergo hepatocellular carcinoma (HCC) surveillance and endoscopic surveillance for varices per standard recommendations. | I, B |
| Hepatotoxic drugs should be used with caution in children with chronic HCV infection after assessment of potential risks versus benefits of treatment. Use of corticosteroids, cytotoxic chemotherapy, and/or therapeutic doses of acetaminophen are not contraindicated in children with chronic HCV infection. | II, C |
| Solid organ transplantation and bone marrow transplantation are not contraindicated in children with chronic HCV infection. | II, C |
| Anticipatory guidance about the potential risks of ethanol for progression of liver disease is recommended for adolescents with chronic HCV infection and their families. Abstinence from alcohol and interventions to facilitate cessation of alcohol consumption, when appropriate, are advised for all persons with chronic HCV infection. | I, C |
Liver disease due to chronic HCV infection generally progresses slowly in children, and cirrhosis and liver cancer occur infrequently. Although elevated serum aminotransferase levels are often noted, HCV-infected children younger than 3 years virtually never develop advanced liver disease.
The initial assessment of children with chronic HCV infection includes exclusion of other causes of liver disease, assessment of disease severity, and detection of extrahepatic manifestations. Testing for concomitant HBV (HBsAg, anti-HBc, and anti-HBs), HIV (anti-HIV), and immunity to HAV (anti-HAV IgG) are recommended due to shared risk factors and the need to vaccinate nonimmune children who may not have received routine childhood HAV and HBV vaccines.
Disease staging in children can be accomplished via physical examination and assessment of routine laboratory parameters including albumin, serum hepatic aminotransferase levels, total bilirubin, international normalized ratio (INR), and platelet count every 6 to 12 months. Serum fibrosis markers also hold promise to stratify disease severity but require further validation (Nielsen, 2019); (Pokorska-Spiewak, 2017); (Mack, 2012). Of note, serum aminotransferase levels are not consistently reflective of disease severity in children. In one study, nearly 33% of children had normal aminotransferase levels despite substantial necroinflammation on biopsy (Casiraghi, 2004).
For children in whom advanced liver disease is a concern, liver imaging to evaluate for splenomegaly or venous collaterals is recommended initially, using liver ultrasound instead of CT or MRI due to its widespread availability and lack of ionizing radiation. Although liver biopsy is considered the gold standard regarding the grade of inflammation and stage of fibrosis, sampling artifact is problematic and most patients and practitioners prefer noninvasive alternatives, such as liver elastography, to determine the presence/absence of cirrhosis, particularly in children. Ultrasound-based liver elastography in children requires the use of specialized probes and cutoff values for advanced fibrosis/cirrhosis that differ from those used in adults, but this approach appears promising for monitoring children with chronic HCV infection (Behairy, 2016); (Geng, 2016); (Lee, 2013).
Due to the slow rate of fibrosis progression among children, there are few, if any, established bona fide risk factors for disease progression. Development of advanced liver disease in children is infrequent until more than 30 years of infection (Jhaveri, 2011); (Goodman, 2008); (Minola, 2002). However, as in adults, children with comorbid disease—such as obesity with nonalcoholic fatty liver disease and congenital heart disease with elevated right heart pressures—and those receiving hepatotoxic drugs should be monitored carefully for disease progression.
Hepatocellular carcinoma (HCC) is rarely encountered among children and has been reported almost exclusively in those with cirrhosis. There are reports that children with chronic HCV infection and a history of childhood leukemia may be at increased risk of developing HCC but evidence is limited (González-Peralta, 2009). In children with cirrhosis, liver ultrasound with or without serum alpha-fetoprotein (AFP) testing every 6 months is recommended for HCC surveillance per AASLD guidelines (Marrero, 2018). A baseline endoscopy is advisable to detect esophageal varices in children with cirrhosis and every 3 years thereafter in the absence of viral clearance. After successful antiviral therapy, the risk for cirrhosis complications decreases substantially.
In children with advanced fibrosis from chronic HCV infection, medications that are known to accelerate hepatic fibrosis (eg, methotrexate) should be avoided, if possible. Similarly, abstinence from alcohol use is strongly advised to minimize disease progression. Although corticosteroids and other immunosuppressants may enhance HCV replication, they are not contraindicated in children with HCV infection and should be prescribed for appropriate indications based on overall risks versus benefits. Of note, icteric flares of HCV—as reported in children and adults with chronic HBV—have not been reported in children receiving an organ transplant or cytotoxic chemotherapy. Although underlying liver disease is a risk factor for development of sinusoidal obstruction syndrome following bone marrow transplantation, the presence of HCV infection should not delay this therapy.
To remain well, untreated children with chronic hepatitis C are encouraged to maintain a healthy body weight due to the known deleterious effects of insulin resistance on fibrosis progression with HCV infection (Kukla, 2015); (Petta, 2011); (Cua, 2008); (Moucari, 2008). Commonly used medications, such as antimicrobial agents, antiepileptics, and cardiovascular agents, should be dosed per standard recommendations. However, nonsteroidal anti-inflammatory drugs and aspirin should be avoided, if possible, in children with cirrhosis and esophageal varices due to concerns of gastrointestinal bleeding and nephrotoxicity. Acetaminophen is a safe and effective analgesic for children with chronic HCV infection when dosed per package insert recommendations.
Recommendations for Whom and When to Treat Among Children and Adolescents With HCV Infection |
|
|---|---|
| RECOMMENDED |
RATING |
| Direct-acting antiviral (DAA) treatment with an approved regimen is recommended for all children and adolescents with HCV infection aged ≥3 years as they will benefit from antiviral therapy, regardless of disease severity. | I, B |
| The presence of extrahepatic manifestations—such as cryoglobulinemia, rashes, and glomerulonephritis—as well as advanced fibrosis should lead to early antiviral therapy to minimize future morbidity and mortality. | I, C |
HCV-related, advanced liver disease is uncommon during childhood. However, liver disease progresses over time with increasing fibrosis severity (Indolfi, 2019); (Mizuochi, 2018); (Bortolotti, 2008); (EPHCVN, 2005); (Resti, 2003). Although uncommon, cirrhosis occurs occasionally in children and adolescents (aged <18 years) with HCV infection. Children have a long life expectancy during which HCV complications may develop. Children and adolescents with HCV infection may also transmit the virus to others.
The high success rates with DAA regimens in adults with chronic HCV infection have been replicated in the pediatric population. Clinical trial data evaluating DAA regimens in children and adolescents have allowed expanded use of these safe, well-tolerated, efficacious HCV therapies in the pediatric population. Treatment of children as young as 12 years is predicted to be very cost-effective with currently approved DAA regimens as well as those in clinical trials (Nguyen, 2019b). Another cost-utility analysis compared DAA treatment at age 6 versus delaying treatment until age 18. The researchers reported the incremental cost-utility ratio for early vs delayed DAA therapy was <$12,000 per QALY gained. They concluded that treatment during early childhood is cost-effective and delaying therapy until early adulthood may result in increased lifetime risk of complications of late-stage liver disease (Greenway, 2019). FDA-approved DAA regimens are available for children aged 3 to <18 years with any genotype of HCV.
|
Recommended regimens listed by pangenotypic, evidence level and alphabetically for Treatment-Naive or Interferon-Experienced Children and Adolescents Without Cirrhosis or With Compensated Cirrhosisa |
||
|---|---|---|
| RECOMMENDED | DURATION |
RATING |
| Combination of glecaprevir/pibrentasvir (weight-based dosing; see Table 1) for children aged ≥ 3 with any genotypeb | 8 weeks | I, B |
| Combination of sofosbuvir/velpatasvir (weight-based dosing; see Table 2) for children ≥3 of age with any genotype | 12 weeks | I, B |
| Combination of ledipasvir/sofosbuvir (weight-based dosing; see Table 3) for children aged ≥3 years with genotype 1, 4, 5, or 6 | 12 weeks | I, B |
|
a Child-Pugh A b A longer duration of therapy (ie, 16 weeks) may be needed for genotype 3 interferon-experienced patients. |
||
|
Recommended regimens listed by pangenotypic, evidence level and alphabetically for DAA-Experienced Children and Adolescents, Without Cirrhosis or With Compensated Cirrhosisa |
||
|---|---|---|
| RECOMMENDED | DURATION |
RATING |
| Genotype 1, 2, 4, 5, or 6: Daily fixed-dose combination of glecaprevir (300 mg)/pibrentasvir (120 mg) for adolescents aged ≥12 years or weighing ≥45 kg with prior exposure to an interferon-based regimen (± ribavirin) and/or sofosbuvir but no exposure to NS3/4A or NS5A protease inhibitors, without cirrhosis | 8 weeks | I, C |
| Genotype 1, 2, 4, 5, or 6: Combination of glecaprevir/pibrentasvir (weight-based dosing; see Table 1) with prior exposure to an interferon-based regimen (± ribavirin) and/or sofosbuvir but no exposure to NS3/4A or NS5A protease inhibitors, with compensated cirrhosisa | 12 weeks | I, C |
| Genotype 3: Combination of glecaprevir/pibrentasvir (weight-based dosing; see Table 1) with prior exposure to an interferon-based regimen (± ribavirin) and/or sofosbuvir but no exposure to NS3/4A or NS5A protease inhibitors, without cirrhosis or with compensated cirrhosisa | 16 weeks | I, C |
| Genotype 1- 6: Combination of glecaprevir/pibrentasvir (weight-based dosing; see Table 1) with prior exposure to NS3/4A protease inhibitors but no NS5A inhibitor exposure, without cirrhosis or with compensated cirrhosisa | 12 weeks | I, C |
| Genotype 1- 6: Combination of glecaprevir/pibrentasvir (weight-based dosing; see Table 1) with prior exposure to an NS5A inhibitor but no NS3/4A protease inhibitor exposure, without cirrhosis or with compensated cirrhosisa | 16 weeks | I, C |
| Genotypes 1-6: Combination of sofosbuvir/velpatasvir (weight-based dosing; see Table 2) with prior exposure to an interferon-based regimen (± ribavirin) and/or sofosbuvir but no exposure to NS3/4A or NS5A protease inhibitors, without cirrhosis or with compensated cirrhosis | 12 weeks | I, C |
| Genotypes 1-6: Combination of sofosbuvir/velpatasvir (weight-based dosing; see Table 2) with weight-based ribavirin (see Table 4) with prior exposure to an interferon-based regimen (± ribavirin) and/or sofosbuvir but no exposure to NS3/4A or NS5A protease inhibitors, with decompensated cirrhosis | 12 weeks | I, C |
| Genotype 4, 5, or 6: Combination of ledipasvir/sofosbuvir (weight-based dosing; see Table 3) for children and adolescents aged ≥3 years with prior exposure to an interferon (± ribavirin) plus an HCV protease inhibitor regimen, without cirrhosis or with compensated cirrhosisa | 12 weeks | I, C |
| Genotype 1: Combination of ledipasvir/sofosbuvir (weight-based dosing; see Table 3) for children and adolescents aged ≥3 years with prior exposure to an interferon (± ribavirin) plus an HCV protease inhibitor regimen, without cirrhosis | 12 weeks | I, C |
| Genotype 1: Combination of ledipasvir/sofosbuvir (weight-based dosing; see Table 3) for children and adolescents aged ≥3 years with prior exposure to an interferon (± ribavirin) plus an HCV protease inhibitor regimen, with compensated cirrhosisa | 24 weeks | I, C |
| a Child-Pugh A | ||
Table 1: Weight-Based Dosing of Glecaprevir/Pibrentasvir for Children Aged ≥3 Years of Age
Body Weight |
Once Daily Dose of Glecaprevir/Pibrentasvir |
|---|---|
| <20 kg | 150 mg/60 mg |
| ≥20 kg to <30 kg | 200 mg/80 mg |
| ≥30 kg to <45 kg | 250 mg/100 mg |
| 45 kg and greater or 12 years of age and older | 300 mg / 120 mg / day |
Table 2: Weight-based dosing for sofosbuvir/velpatasvir fixed dose combination in children ≥ 3 years of age
Body Weight |
Once Daily Dose of Sofosbuvir/Velpatasvir |
|---|---|
| < 17 kg | 150 mg/37.5 mg |
| 17 - < 30 kg | 200 mg/50 mg |
| ≧ 30 kg | 400 mg/100 mg |
Table 3: Weight-Based Dosing of Ledipasvir/Sofosbuvir for Children Aged ≥3 Years
Body Weight |
Once Daily Dose of Ledipasvir/Sofosbuvir |
|---|---|
| <17 kg | 33.75 mg/150 mg |
| 17 to <35 kg | 45 mg/200 mg |
| ≥35 kg | 90 mg/400 mg per day |
Table 4. Weight-Based Dosing of Ribavirin for Children Aged ≥3 Years
Body Weight |
Daily Dose of Ribavirin (divided AM and PM) |
|---|---|
| <47 kg | 15 mg/kg |
| 47 to 49 kg | 600 mg |
| 50 to 65 kg | 800 mg |
| 66 to 80 kg | 1000 mg |
| >80 kg | 1200 mg |
The daily fixed-dose combination of glecaprevir (300 mg)/pibrentasvir (120 mg) was approved for adolescents aged 12 through 17 years in April 2019. In the registration trial, 47 adolescents were treated with the adult-approved coformulated preparation; the duration of treatment was based on viral genotype, prior treatment, and cirrhosis status (Jonas, 2019). Genotypes 1 through 4 were represented in the trial. Two participants were HIV coinfected, none had cirrhosis, and 11 had a prior treatment failure with peginterferon/ribavirin. SVR12 was 100%. The study drugs were well tolerated with no serious adverse events and no drug discontinuations.
Although there are no data from the adolescent population, EXPEDITION-8 evaluated 8 weeks of glecaprevir/pibrentasvir among 343 treatment-naive adults with genotype 1, 2, 3, 4, 5, or 6 and compensated cirrhosis. Overall SVR12 rates were 99.7% (334/335) in the per-protocol population and 97.7% (335/343) in the intention-to-treat population (Brown, 2019). Similarly, FDA approval and HCV guidance panel HCV treatment recommendations for DAA-experienced adolescents are based on clinical trial data from adults (Asselah, 2018b); (Puoti, 2018); (Wyles, 2018); (Zeuzem, 2018); (Forns, 2017).
Part 2 of the DORA trial examined the pharmacokinetics, safety and efficacy of glecaprevir/pibrentasvir among children aged 3 to <12 years with HCV of any genotype who were treatment naive or interferon/ribavirin experienced. Although the trial was designed to include children with compensated cirrhosis, none of the participants had cirrhosis at enrollment. The majority (98%; 78/80) of the children who received glecaprevir/pibrentasvir were treatment naive; a single participant was HIV/HCV coinfected. The overall SVR12 with the optimal drug dosages/ratios was 96%. Of the 2 nonresponders, 1 child discontinued treatment after only 1 dose because of palatability and the other after 4 days due to a drug-related rash. No clinically significant laboratory abnormalities or liver-related toxicities were observed (Jonas, 2021). This regimen was approved for use in children 3 to < 12 years of age in 2021.
Given its pangenotypic activity, safety, and efficacy record in adult patients, glecaprevir/pibrentasvir is recommended as a first choice for pediatric and adolescent HCV treatment. As in adults, coadministration of carbamazepine, efavirenz-containing regimens, and St. John’s wort is not recommended since these compounds may decrease concentrations of glecaprevir and pibrentasvir.
The efficacy of sofosbuvir/velpatasvir once daily for 12 weeks was evaluated in an open-label trial among 173 pediatric participants aged ≥6 years with genotype 1, 2, 3, 4, or 6 infection, without cirrhosis or with compensated cirrhosis. Eighty-five percent of participants (147/173) were treatment naive and 15% (26/173) were treatment experienced. Overall SVR12 was ≥92% across genotypes (Jonas, 2019a).
Among 102 adolescents aged 12 to <18 years, 78% (n=80) were treatment naive and 22% (n=22) were treatment experienced. The median age was 15 years (range 12 to 17 years); 51% were female. The genotype distribution among the participants was 74% genotype 1, 6% genotype 2, 12% genotype 3, 2% genotype 4, and 6% genotype 6. No adolescents had known cirrhosis. The majority (89%; 91/102) had been infected through vertical transmission. SVR12 rates were 93% in adolescents with genotype 1, 91% in those with genotype 3, and 100% in participants with genotype 2, 4, or 6. One participant discontinued treatment at week 4 and subsequently relapsed. The other 4 participants who did not achieve SVR12 did not meet virologic failure criteria (lost to follow-up).
Among 71 children aged 6 to <12 years, the genotype distribution was 76% genotype 1, 3% genotype 2, 15% genotype 3, and 6% genotype 4. None of the participants had known cirrhosis. Ninety-four percent (n=67) were treatment naive and 6% (n=4) 4 were treatment experienced. The median age was 8 years (range 6 to 11 years); 54% were female. The majority of children (94%; 67/71) had been infected through vertical transmission. SVR12 rates were 93% (50/54) in children with genotype 1, 91% (10/11) in those with genotype 3, and 100% in participants with genotype 2 (2/2) or genotype 4 (4/4). One participant had on-treatment virologic failure; the other 4 participants who did not achieve SVR12 did not meet virologic failure criteria (lost to follow-up).
Sofosbuvir/velpatasvir was approved by the FDA for pediatric patients aged ≥6 years in March 2020 and for children 3 to < 6 years of age in June 2021. Given its pangenotypic activity, safety, and efficacy, sofosbuvir/velpatasvir is recommended as a first choice for HCV treatment in children and adolescents. Due to reports from experience among adults, coadministration of sofosbuvir/velpatasvir with amiodarone is not recommended due to the risk for symptomatic bradycardia.
Ledipasvir/sofosbuvir is approved for use in children aged 3 through 17 years with genotype 1, 4, 5, or 6 infection. In a phase 2, multicenter, open-label study of 100 adolescents with genotype 1 treated for 12 weeks with the adult formulation of ledipasvir/sofosbuvir, SVR12 was documented in 98% of participants (Balistreri, 2017). The 2 patients who did not achieve SVR12 were lost to follow-up during or after treatment. Eighty percent of the patients were treatment naive. One patient had cirrhosis, 42 did not, and the cirrhosis status was unknown in the remaining 57. The regimen was safe and well tolerated in this population, and the adult dosage formulation resulted in pharmacokinetic characteristics similar to those observed in adults. Two clinical trials supporting the approval of ledipasvir/sofosbuvir in the pediatric population aged 3 through 11 years demonstrated high SVR12 rates comparable to those seen in adults (Schwarz, 2019); (Murray, 2018). Among children <12 years of age, dosing is weight based (see Table 1). Twelve weeks of ledipasvir/sofosbuvir is recommended for treatment-naive children and adolescents aged ≥3 years without cirrhosis or with compensated cirrhosis (Child-Pugh A). This regimen is also recommended for interferon-experienced (± ribavirin, with or without an HCV protease inhibitor) children and adolescents aged ≥3 years with genotype 1 or 4. A 12-week course is recommended for patients without cirrhosis; 24 weeks is recommended for those with compensated cirrhosis.
In September 2019, the FDA approved weight-based sofosbuvir plus ribavirin (see Table 4) for treatment-naive or interferon-experienced (± ribavirin) children aged ≥3 years with genotype 2 or 3, without cirrhosis or with compensated cirrhosis (Child-Pugh A). This regimen is no longer favored because pangenotypic ribavirin-free treatments are now available for children as young as 3 years of age.
People who inject drugs (PWID), men who have sex with men (MSM), and individuals in jails and prisons bear a particularly high burden of chronic HCV infection. Injection drug use accounts for the majority of new HCV infections, and the rising opioid epidemic has become an important force in the perpetuation of the HCV epidemic. Acute HCV infection is also increasingly being reported among HIV-infected and
Achieving the goal of HCV elimination will depend on diagnosing HCV and treating HCV infection in these groups, and implementing harm reduction strategies to prevent future infections. As a result, the panel has chosen to focus attention on HCV management among these key populations to reduce HCV transmission and decrease HCV-related morbidity and mortality. The first subsection of the key populations guidance focuses on recommendations for HCV testing, treatment, and harm reduction among PWID. The second subsection focuses on testing, treatment, and prevention of HCV among MSM. The final subsection provides recommendations for screening and treatment of HCV in jail and prison settings. Chronic HCV cannot be eliminated without implementation of strategies to reach these populations, and the recommendations in these subsections provide guidance in this effort.
The following subsections include guidance for management of patients with HCV in key populations.
Injection drug use (IDU) is the most common risk factor for HCV infection in North America and Europe, with an HCV seroprevalence of 18% to 88% depending on geographic location (Degenhardt, 2017) and duration of IDU exposure (Mateu-Gelabert, 2022); (Amon, 2008). In this section, the term people who inject drugs (PWID) includes individuals who are actively using drugs and those who have previously used injection drugs.
The first few years after an individual begins to inject drugs constitute a high-risk period during which the rate of HCV infection can exceed 40% (Maher, 2006). According to the National Survey on Drug Use and Health, heroin use has increased across the US among men and women, most age groups, and all income levels (Jones, 2015). IDU accounts for the majority of new HCV infections (approximately 70%) and is the driving force in the perpetuation of the epidemic. Given these facts and the absence of a vaccine against HCV, testing and linkage to care combined with antiviral treatment have the potential to decrease HCV incidence and prevalence (NAS, 2017); (Martin, 2013).
Recommendations for Screening and Treatment of HCV Infection in People Who Inject Drugs (PWID) |
|
|---|---|
| RECOMMENDED |
RATING |
| Annual HCV testing is recommended for PWID with no prior testing, or past negative testing and subsequent injection drug use. Depending on the level of risk, more frequent testing may be indicated. | IIa, C |
| Substance use disorder treatment programs and needle/syringe exchange programs should offer routine, opt-out HCV-antibody testing with reflexive or immediate confirmatory HCV-RNA testing and linkage to care for those who are infected. | IIa, C |
| PWID should be counseled about measures to reduce the risk of HCV transmission to others. | I, C |
| PWID should be offered linkage to harm reduction services including intranasal naloxone, needle/syringe service programs, medications for opioid use disorder, and other substance use disorder treatment programs. | I, B |
| Active or recent drug use or a concern for reinfection is not a contraindication to HCV treatment. | IIa, B |
All individuals who currently inject drugs or have previously used injection drugs should be tested for HCV infection. Data are limited regarding the optimal interval for repeat testing among individuals actively using drugs. An HCV-antibody test is recommended and if the result is positive, current infection should be confirmed by immediate HCV-RNA testing (see HCV Testing and Linkage to Care). This can be accomplished using phlebotomy for a combined reflex test performed by a laboratory, which is appropriate for clinical settings. In certain community settings, a point-of-care antibody test with an immediate blood draw or dried blood spot collection for a confirmatory HCV-RNA test may be implemented.
Among persons at risk for HCV reinfection after previous spontaneous or treatment-related viral clearance, HCV-RNA testing is recommended because an HCV-antibody test is expected to remain positive. Among persons with a negative HCV-antibody test who are at high risk for a new HCV infection due to current IDU, testing for HCV RNA or follow-up testing for HCV antibody is recommended if HCV exposure may have occurred within the past 6 months.
Integration of HCV testing services into substance use disorder treatment programs, needle/syringe service programs, and acute detoxification programs provide an opportunity for routine screening in this key population (Aronson, 2017); (Harris, 2010).
Treatment of HCV-infected PWID should ideally be delivered in a multidisciplinary care setting with services to reduce reinfection risk and manage the common social and psychiatric comorbidities in this population.
Regardless of the treatment setting, recent and active IDU are not absolute contraindications to HCV therapy. There is strong evidence from various settings in which PWID have demonstrated adherence to treatment and low rates of reinfection, countering arguments that have been commonly used to limit HCV therapy access in this patient population (Coffin, 2019); (Dore, 2016); (Hellard, 2014); (Aspinall, 2013); (Grebely, 2011). Modeling studies illustrate the high return on the modest investment of addressing this often-ignored segment of the HCV-infected population (Barbosa, 2019); (Fraser, 2018b); (Zelenev, 2018); (Martin, 2013b). Conversely, there are no data to support the utility of pretreatment screening for illicit drug or alcohol use in identifying a population more likely to successfully complete HCV therapy. These requirements should be abandoned because they create barriers to treatment, add unnecessary cost and effort, miss an opportunity to decrease HCV transmission, and potentially exclude populations that are likely to obtain substantial benefit from therapy. Instead, scaling up HCV treatment in PWID is necessary to positively impact the HCV epidemic in the US and globally.
Recent hepatitis C test-and-link programs have identified the use of patient navigators or care coordinators to be an important intervention in overcoming challenges to linkage to and retention in care (Coyle, 2019); (Ford, 2017); (Coyle, 2016); (Ramirez, 2016); (Coyle, 2015); (Trooskin, 2015). The Check Hep C program in New York City compared services delivered at 2 clinical care sites to 2 sites that linked patients to off-site care. Participants receiving clinical care co-located with testing services had higher odds of initiating treatment than those linked to off-site care (Ford, 2017). Ongoing assessment of efficacy and comparative effectiveness of this and additional strategies is a crucial area of future research for patients with chronic HCV. Replication and expansion of best practices and new models for linkage to HCV care will be essential to maximize the public health impact of newer HCV treatment paradigms.
Clinical trials among PWID reporting current IDU at the start of HCV treatment and/or continued use during therapy demonstrate SVR12 rates approaching 95% (Grebely, 2018); (Dore, 2016). Moreover, high SVR rates among PWID are not limited to clinical trials but are also observed in clinical practice settings. A cohort study was conducted with 89 patients initiating HCV treatment between January 2014 and August 2015 at a primary care clinic in the Bronx, New York. Four patient groups were compared: no active drug use or medications for opioid use disorder (MOUDs); no active drug use with MOUDs; active drug use without MOUDs; and active drug use MOUDs. The study found that regardless of active drug or MOUD use, patients who received direct-acting antiviral (DAA) therapy at this urban primary care clinic achieved high HCV cure rates (SVR ≥95%) (Norton, 2017).
Dispensing DAA therapy within a program that provides MOUDs increases the likelihood of PWID engagement in HCV treatment (Falade-Nwulia, 2019). Importantly, MOUDs do not compromise HCV treatment outcomes. Similar SVR12 rates are achieved by PWID engaged in MOUD use compared with individuals not engaged with such medications in clinical trials and cohort studies of various DAA regimens (Macías, 2019); (Dore, 2016); (Grebely, 2016); (Lalezari, 2015); (Zeuzem, 2015); (Feld, 2014). HCV-infected patients receiving MOUDs who were treated with elbasvir/grazoprevir had high rates of adherence to antiviral treatment and SVR12 rates >89% regardless of ongoing IDU (Dore, 2016). Similarly, an SVR12 of 97.4% was reported in a clinical trial evaluating ombitasvir/paritaprevir/ritonavir plus dasabuvir and ribavirin for 12 weeks among patients receiving MOUDs (Lalezari, 2015). Further, an analysis of a clinical trial evaluating outcomes of sofosbuvir/velpatasvir treatment in patients receiving MOUDs (n=51) compared to those not receiving these medications (n=984) demonstrated that MOUD use did not significantly reduce treatment completion, antiviral adherence, SVR12, or safety (Grebely, 2016).
Optimal models of HCV treatment among patients receiving MOUDs are still being evaluated. A recent trial conducted among PWID receiving MOUDs within 3 New York programs suggested that directly observed DAA therapy was associated with greater antiviral adherence than self-administered individual DAA treatment (86% versus 75%; p=0.001) (Akiyama, 2019). Importantly, opioid IDU and sharing has been observed to decrease following DAA HCV treatment (Artenie, 2020).
Recommendation for Testing for Reinfection in People Who Inject Drugs (PWID) |
|
|---|---|
| RECOMMENDED |
RATING |
| At least annual HCV-RNA testing is recommended for PWID with recent injection drug use after they have spontaneously cleared HCV infection or have been successfully treated. | IIa, C |
As HCV therapy is expanded to populations of PWID with high-risk behaviors for re-exposure, acknowledgement that HCV reinfection will occur in some individuals is critical, and appropriate strategies must be in place to maximize prevention of reinfection and offer retreatment for reinfection (Grebely, 2017). Importantly, the rate of HCV reinfection in the PWID population is lower (2.4/100 person-years) than the rate of incident HCV infection in the general population of PWID (6.1 to 27.2/100 person-years), although the rate of reinfection increases with active or ongoing IDU (up to 7.4/100 person-years) (Akiyama, 2019b); (Aspinall, 2013); (Grady, 2013).
Data suggest that reinfection is rare in drug users who clear HCV with therapy even if they continue to inject drugs provided steps are taken to minimize the risk. Studies of HCV reinfection in PWID have demonstrated rates of reinfection post SVR ranging from 1 to 5/100 person-years in patients who have ever injected drugs, increasing to 3 to 33/100 person-years in patients with continued injecting risk behavior (Midgard, 2016b); (Marco, 2013); (Grady, 2012); (Grebely, 2012); (Bate, 2010); (Grebely, 2010); (Currie, 2008); (Dalgard, 2002). Relapse into drug use has been associated with HCV reinfection after cure (Midgard, 2016b) while interventions that reduce drug use, such as utilization of MOUDs and mental health services, have been associated with reduced HCV reinfection risk (Islam, 2017). These services should be made available to PWID.
PWID found to be HCV reinfected should be retreated. Retreatment of a new reinfection should be as detailed in the Initial Treatment section. Increasing the HCV treatment rate among the PWID population would reduce numbers of new HCV and liver-related disease cases (Jiang, 2017). In a study that evaluated reinfection and injecting risk behavior following DAA therapy, participants on MOUDs for ≥3 months had a reinfection rate of 2.3/100 person-years, with a persistent reinfection rate of 1.6/100 person-years due to spontaneous HCV clearance in several instances. A reinfection rate of 4.2/100 person-years was found among those who reported IDU (Dore, 2017).
Harm reduction is a way of preventing disease and promoting health that meets people where they are, and provides the tools and information they need to keep themselves and those around them well (Logan, 2010). Harm reduction places drug use within the larger sociopolitical spheres of poverty, criminalization, and mental health. Accepting that not everyone is ready or able to curtail or stop high-risk behavior, harm reduction focuses on promoting a spectrum of scientifically proven, practical strategies for reducing the negative consequences of drug use and other high-risk behaviors. Harm reduction strategies include but are not limited to condom distribution; access to sterile injection equipment; utilization of MOUDs (such as methadone, buprenorphine and naltrexone); safe injection spaces; and overdose education and naloxone distribution. Heroin overdose deaths in the US increased 286% from 2002 to 2013 (Jones, 2015). Broad implementation of harm reduction strategies has the potential to significantly impact the HCV epidemic.
Medications for Opioid Use Disorder
Methadone, buprenorphine, and naltrexone are FDA-approved treatments for opioid use disorder with evidence from randomized controlled trials and real-world cohorts to support their effectiveness in reducing opioid use, improving mortality, decreasing criminal activity, and improving social functioning and retention in care (Tasillo, 2017); (Kampman 2015); (Volkow, 2014). Methadone is a long-acting opioid agonist that has the longest history in clinical use and is proven to reduce illicit drug use and improve social functioning (Mattick, 2009). Although methadone is effective, concern about diversion leads to methadone maintenance being highly regulated in the US, typically requiring daily visits to a dedicated dispensing clinic (Mattick, 2014). Buprenorphine-naloxone is a partial opioid agonist that also relieves withdrawal, and quells opioid craving. Multiple randomized trials support its effectiveness in reducing drug use and improving retention in care (Tasillo, 2017); (Volkow, 2017); (Kampman, 2015); (Volkow, 2014); (Mattick, 2014); (Moore, 2012); (Weiss, 2011); (Comer, 2010); (Jones, 2010); (Ling, 2010); (Lucas, 2010); (Mattick, 2009); (Kakko, 2007); (Fischer, 2006); (Jones, 2005); (Fudala, 2003); (Kakko, 2003); (Johnson, 2000); (Ling, 1998); (O’Connor, 1998); (Ling, 1996); (Johnson,1995). Buprenorphine-naloxone’s major benefits include that it is a partial agonist which limits its overdose risk; coformulation with naloxone provides a deterrent from injecting; and it can be successfully prescribed in routine primary care settings (Korthuis, 2017); (LaBelle, 2016); (Fudala, 2003). Prescribing buprenorphine-naloxone requires 8 hours of training and registration with the US Drug Enforcement Agency and receiving a waiver from the Substance Abuse Mental Health Services Administration, which limits the number of providers (Stein, 2015). Naltrexone is an opioid antagonist that prevents the euphoric and respiratory effects of opioids, reducing cravings (SAMHSA, 2020). Naltrexone has low diversion potential and requires no special licensing for prescribers (Rudd, 2016). Further, it is available as a monthly injection. Naltrexone precipitates opioid withdrawal, however, and is therefore only initiated in opioid-abstinent patients.
Several reviews have identified MOUDs as effective in reducing illicit opioid use (Mattick, 2014); (Mattick, 2009) and opioid-related death and all-cause mortality (Sordo, 2017); (Degenhardt, 2009), and improving quality of life (Lawrinson, 2008); (Ward, 1999). Participation in methadone maintenance treatment has been shown to be protective against hepatitis C incidence among PWID, with a dose-response protective effect with increasing methadone exposure on hepatitis C incidence (Nolan, 2014).
Syringe Service Programs
Syringe service programs (SSPs) were developed to reduce the spread of bloodborne diseases among injection drug users. These programs provide PWID with sterile syringes and other equipment (cookers, filters, sterile water, alcohol swabs) to reduce the risk of bloodborne disease (eg, HIV and HCV) transmission associated with sharing injection equipment. These programs were developed in the 1980s and often include drug treatment referrals, peer education, and HIV prevention. Areas with greater syringe access through SSPs have lower rates of hepatitis C among PWID. A prospective study of PWID in New York City found a significant decline in HCV rates from 1990 to 2001, corresponding to an increase in the number of syringes distributed by SSPs during this period (Des Jarlais, 2005).
Overdose Education and Naloxone Distribution (OEND)
HCV treatment is a touchpoint with the care delivery system and should be used as an opportunity to mitigate the harms of drug use, especially overdose risk. Naloxone is a powerful opioid antagonist that reverses the respiratory depressive effects of opioids and is lifesaving to those experiencing opioid overdose (Wermeling, 2015). Expanding access to intranasal naloxone significantly decreases mortality at the community level (Walley, 2013). Many states have standing orders for intranasal naloxone, which allow providers to dispense naloxone directly to patients. When no standing order exists or when it is not feasible to provide naloxone directly, providers should offer patients a prescription for naloxone to fill at a local pharmacy. Importantly, naloxone is not an opioid and carries no overdose risk, no dependency risk, and no risk of diversion. Naloxone is safe and effective and can be prescribed with confidence by HCV providers who do not treat addictions more generally.
Persons cured of chronic HCV no longer transmit the virus to others. As such, successful HCV treatment benefits public health. Several health models have shown that even modest increases in successful HCV treatment among PWID can decrease prevalence and incidence (Hellard, 2014); (Martin, 2013); (Martin, 2013b); (Durier, 2012). Models developed to estimate the impact of HCV testing and treatment on the burden of HCV at a country level reveal that large decreases in HCV prevalence and incidence are possible as more persons are successfully treated (Martin, 2015); (Wedemeyer, 2014). Elimination of HCV among PWID will also require scaling up harm reduction services (Fraser, 2018).
Several outbreaks of sexually transmitted HCV infection among HIV-infected men who have sex with men (MSM) have been reported since 2000 (Wandeler, 2012); (van de Laar, 2010); (Urbanus, 2009); (Matthews, 2007). A recent systematic review reported an HCV incidence of 6.35/1000 person-years among HIV-infected MSM (Jin, 2017). The determinants of sexually transmitted, incident HCV among HIV-positive MSM have not been thoroughly characterized but risk factors have been identified. Group sex practices that can cause trauma to rectal mucosal tissue (eg, receptive anal intercourse without a condom and receptive fisting) and rectal bleeding are associated with HCV transmission among HIV-infected MSM (Daskalopoulou, 2017); (Page, 2016); (Apers, 2015); (Vanhommerig, 2015); (Witt, 2013); (Wandeler, 2012); (CDC, 2011); (Schmidt, 2011); (Danta, 2007).
The recent proliferation of chemsex (also known as party and play [PNP])—use of crystal methamphetamine, mephedrone, or gamma-hydroxybutyrate, sometimes with phosphodiesterase type 5 inhibitors (which lowers inhibitions, creates feelings of invulnerability, increases stamina, and inhibits ejaculation) before or during sex—has also been associated with incident HCV infection (Pufall, 2018); (Hegazi, 2017); (NHS, 2014). These HCV infections have been occurring especially in men who already have ulcerative and rectal sexually transmitted infections including syphilis, lymphogranuloma venereum, and genital herpes (Bottieau, 2010); (van de Laar, 2007); (Gambotti, 2005); (Gotz, 2005); (Browne, 2004); (Ghosn, 2004).
While it is not completely clear why higher rates of incident HCV have been reported in HIV-infected compared to uninfected MSM, behavioral factors such as serosorting (sex between partners of the same HIV status with the aim of minimizing HIV transmission risk) and increased rates of anal sex without condoms by HIV-infected men have been implicated (Mao, 2011). In a recent study of 33 HIV/HCV-coinfected MSM, one-third shed HCV in their semen (Turner, 2016). In addition to being found in semen, rectal shedding of HCV has also been reported in HIV/HCV-coinfected MSM (Foster, 2017b).
Acute HCV infections have been recently reported among HIV-uninfected MSM who present for pre-exposure prophylaxis (PrEP) (Hoornenborg, 2017). These HIV-uninfected men became infected with HCV strains known to be circulating in HIV sexual transmission networks. Thus, there is growing concern that with the implementation of PrEP, high-risk HIV-uninfected MSM may be at increased risk of incident HCV through unprotected sexual intercourse with HCV-infected MSM. The risk factors for acute HCV infection in these patients remain unknown but may be similar to those reported in HIV-infected MSM.
Recommendations for Testing and Prevention of HCV Infection in Men Who Have Sex With Men (MSM) |
|
|---|---|
| RECOMMENDED |
RATING |
| Annual HCV testing is recommended for sexually active HIV-infected adolescent and adult MSM. Depending on the presence of high-risk sexual or drug use practices, more frequent testing may be warranted. | IIa, C |
| HCV testing at HIV pre-exposure prophylaxis (PrEP) initiation and at least annually thereafter (while on PrEP) is recommended in HIV-uninfected MSM. Depending on sexual or drug use risk practices, more frequent testing may be warranted. | IIa, C |
| All MSM should be counseled about the risk of sexual HCV transmission with high-risk sexual and drug use practices, and educated about measures to prevent HCV infection or transmission. | IIa, C |
Practitioners treating HIV-infected adolescent and adult MSM should be on high alert for acute HCV infection, which is most often asymptomatic (see the HCV in Children section). In accordance with US Centers for Disease Control and Prevention sexually transmitted diseases (STDs) screening recommendations, HCV screening should be performed at least annually and may be done more frequently, depending on the presence of local and individual factors such as high HCV prevalence and/or incidence locally, high-risk sexual behavior (eg, unprotected [by a condom] receptive anal intercourse, group sex, fisting, chemsex), and ulcerative STD(s) or STD-related proctitis (Pufall, 2018); (Daskalopoulou, 2017); (Page, 2016); (Apers, 2015); (CDC, 2015); (Vanhommerig, 2015); (NHS, 2014); (Witt, 2013); (Wandeler, 2012); (CDC, 2011); (Schmidt, 2011); (Bottieau, 2010); (Danta, 2007); (van de Laar, 2007); (Gambotti, 2005); (Gotz, 2005); (Browne, 2004); (Ghosn, 2004).
Screening should be performed using an HCV-antibody test in most instances. However, individuals with self-reported recent high-risk exposures and/or newly elevated alanine aminotransferase (ALT) levels should have HCV screening with both HCV-antibody and HCV-RNA tests due to concern for acute HCV infection. Those found to be chronically HCV infected should be offered antiviral treatment to prevent liver disease progression and transmission to others. These patients should also be counseled about risk factors for HCV transmission and the potential for HCV reinfection after cure (Ingiliz, 2017); (Ingiliz, 2014); (Lambers, 2011). Subsequent care for acute HCV should be as detailed in the Management of Acute HCV section.
To reduce the risk of sexually transmitted HCV and other STDs, MSM should be counseled to use condoms with all sex acts. They should also be informed about the high risk of HCV transmission associated with sharing any equipment used for preparing and injecting or snorting drugs. If indicated (and available), providers should offer referrals to syringe service programs and culturally competent counseling/drug treatment, and encourage patients to seek testing for sexually transmitted infections if they have been at risk. Among patients who are using opioids, discussion of preventing HCV infection is also an opportunity to provide opioid education and naloxone distribution (OEND), which is an effective intervention to prevent overdose death.
Although PrEP can prevent sexual transmission of HIV, it is not protective against HCV or other sexually transmitted infections. HIV-uninfected MSM who present for PrEP should receive risk reduction counseling. HIV-uninfected MSM on PrEP should also receive at least annual HCV screening for identification of incident infections.
Recommendation on Treatment of HCV in Men Who Have Sex With Men (MSM) |
|
|---|---|
| RECOMMENDED |
RATING |
| Antiviral treatment for HCV-infected MSM should be coupled with ongoing counseling about the risk of HCV reinfection, and education about methods to reduce HCV reinfection risk after cure. | I, B |
Because MSM may be at high risk of transmitting HCV to others, HCV infection should be treated both for individual benefit and to prevent HCV transmission. HIV-infected MSM are considered an important population for HCV elimination through treatment as prevention (Martin, 2015). The population-level benefit of expansion of HCV treatment in populations of HIV-infected MSM has been evaluated in modeling studies (Martin, 2016); (Salazar-Vizcaya, 2016). Additionally, real-world data support the potential for HCV treatment as prevention in cohorts of HIV/HCV-coinfected MSM. Analysis of data from the Dutch acute HCV in HIV study group (DAHHS) showed a 50% reduction in acute HCV incidence between 2014 and 2016 within 1 year of expansion of HCV therapy through unrestricted direct-acting antiviral (DAA) availability to HIV-infected MSM (Boerekamps, 2017).
HCV treatment should be coupled with education addressing the potential for HCV reinfection and risk factors for transmission to reduce the risk of transmission to others and subsequent reinfection after HCV cure. Brief counseling interventions delivered in clinical settings have been shown to reduce HIV transmission risk and may be effective in reducing HCV transmission risk (Boerekamps, 2017); (Myers, 2010); (Richardson, 2004).
Recommendation on Prevention of HCV Reinfection in Men Who Have Sex With Men (MSM) |
|
|---|---|
| RECOMMENDED |
RATING |
| At least annual (and risk-based, if indicated) HCV testing with HCV RNA is recommended for sexually active MSM after successful treatment or spontaneous clearance of HCV infection. | IIa, C |
High HCV reinfection rates, ranging from 7.3 to 15.2/100 person-years, have been reported after HCV treatment and cure among HIV-infected MSM (Ingiliz, 2017); (Martin, 2015b); (Lambers, 2011). In an analysis of 606 MSM from 8 centers in Europe, an increase in HCV reinfection incidence rates was reported with each subsequent reinfection (HCV reinfection incidence 7.3/100 person-years for the first reinfection and 18.8/100 person-years for the second reinfection) (Ingiliz, 2017). For this reason, it is important to provide patients with clear, nonjudgmental, accurate information about reducing their risk for sexually transmitted HCV. This counseling should be ongoing. Additionally, clinicians should monitor and test for HCV reinfection in sexually active MSM after cure, regardless of HIV status. Individuals found to be HCV reinfected should be retreated. HCV treatment in this setting should be as detailed in the Initial Treatment of HCV section.
HCV infection disproportionately affects individuals in correctional institutions, which include jails (short-stay facilities that typically house persons for sentences of up to 1 year) and prisons (long-term facilities for persons with a felony conviction). Recent cross-sectional surveys suggest that HCV seroprevalence among incarcerated populations in the US ranges from 3.0% to 34.6% (Busschots, 2022), which exceeds the 1.7% HCV seroprevalence in the general population (Hofmeister, 2019). However, HCV prevalence in correctional populations is not geographically uniform and can vary by state and region (Varan, 2014). Injection drug use is the most common risk factor for HCV transmission in correctional settings (Ruiz, 1999); (Spaulding, 2006). HCV-associated liver disease is a frequent cause of death in inmates and has recently surpassed death from HIV (Spaulding, 2011); (Spaulding, 2015).
Approximately 30% of all persons with HCV infection in the US spend at least part of the year in a correctional institution (Hammett, 2002); (Varan, 2014). Unfortunately, most HCV-infected individuals in correctional facilities are unaware of their infection (Spaulding, 2012). Given the high prevalence of HCV infection in correctional settings coupled with the fact that more than 10 million individuals pass through jails and prisons each year, as many as 1 million persons with undiagnosed HCV infection might come into contact with the correctional system each year (Spaulding, 2012); (Rich, 2014). More than 90% of these individuals are eventually released and re-enter the general population, where they can contribute to HCV spread in the community (Macalino, 2004); (Rich, 2014) and may have little contact with the healthcare system (Fox, 2005); (Bushway, 2006); (Rich, 2014b); (Neate, 2016). Moreover, 68% of prisoners are reincarcerated for a new crime within 3 years of their release from prison (Durose, 2014). Recidivism can further promote the spread of HCV within correctional settings.
Both the US Preventive Services Task Force and the World Health Organization recommend that all incarcerated persons undergo HCV testing (WHO, 2016); (Moyer, 2013b). Despite these recommendations and the high prevalence of HCV infection in correctional institutions, HCV testing is not universally performed in this setting.
HCV testing and treatment have been historically uncommon in jails, primarily because of the short duration of incarceration and lack of available resources (Maurer, 2015). With approximately 11 million jail admissions annually (Minton, 2016), jails represent an important public health setting in which to test for HCV infection and treat persons with chronic HCV.
Jails have also not had the resources and systems to enable continuation of community-initiated HCV therapy. If detainees are unable to continue HCV treatment while incarcerated in jail, the interruption in therapy will adversely affect the likelihood of achieving a cure and could promote development of viral resistance. Without systems to facilitate continuation of antiviral therapy, jails may interfere with community HCV treatment efforts and societal payers will suffer losses on investments.
The bulk of the evidence on current HCV testing and treatment in the prison setting is based on a 2015 national survey conducted by the American Correctional Association and the Coalition of Correctional Health Authorities research and health outcomes working group (Maurer, 2015). According to this survey, some type of HCV testing is performed in the majority of prisons but routine opt-out testing is generally not conducted across the prison system. Additionally, there are major differences in approaches to HCV testing and prevention counseling. The most common triggers for HCV testing in a prison setting were physician request, identified risk factors, and inmate request. Only 16% of prison facilities tested all inmates with an HCV-antibody test upon entry. Selection of patients for antiviral therapy also varied across prison systems. The survey found that antiviral therapy for chronic HCV was available in 90% of prisons. However, few inmates actually received treatment, primarily due to antiviral therapy expense and lack of availability of trained staff. Moreover, despite the fact that injection drug use was the major risk factor for HCV transmission in this population, only half of the prison facilities combined substance use disorder treatment with HCV therapy.
More recently, investigators at Yale University administered a survey to the directors of the departments of corrections in all 50 US states that inquired about current HCV practices within state correctional facilities (Beckman, 2016). This survey included questions about the number of inmates in the state’s prisons known to be HCV infected on or about December 31, 2014; the number of prisoners receiving any form of HCV treatment at that time; and the availability of relevant resources for inmates with known HCV infection. Representatives from 41 states completed the questions on the number of inmates with chronic HCV infection and the proportion receiving antiviral treatment. The overall number of inmates who were reported to have chronic HCV in the 41 reporting states was 106,266 prisoners, corresponding to 10% of the overall prison population in these states. Among these inmates, only 0.89% (n=949) received any form of HCV treatment on or about December 31, 2014. States used a variety of factors to prioritize HCV treatment among inmates, particularly cirrhosis, sentence length, likelihood of recidivism, potential for antiviral adherence, and chance of HCV reinfection. States with a relatively high proportion of inmates reported to have HCV infection did not treat a greater number of patients than states with a lower proportion of infections.
Representatives from 49 of the state departments of corrections completed the questions on resources related to HCV infection. Seventeen states reported offering routine opt-out HCV testing of inmates. Among the 32 states without routine opt-out HCV testing, the main indications for HCV testing were abnormal results from other tests, HIV infection, or a substance use disorder. Medication-assisted treatment programs for substance use disorders were available through 14 state departments of corrections. Four states reported that they followed all of the Federal Bureau of Prisons guidelines (FBP, 2016).
Given the high prevalence of HCV among persons in the US correctional system, the success of the national HCV elimination effort will depend on identifying chronically infected individuals in jails and prisons, linking these persons to medical care for management, and providing access to antiviral treatment (NAS, 2017). Diagnosis of chronic HCV in correctional settings followed by linkage to care and successful antiviral treatment can ultimately reduce the risk of liver-related and extrahepatic complications, and has the potential to decrease HCV transmission in correctional facilities and the community after release (van der Meer, 2012); (Harris, 2016); (He, 2016).
Recommendations for Screening and Treatment of HCV Infection in Jails |
|
|---|---|
| RECOMMENDED |
RATING |
Jails should implement opt-out HCV testing consisting of HCV-antibody testing followed by confirmatory HCV-RNA testing if antibody-positive.
|
IIa, C |
Recommendations for Screening and Treatment of HCV Infection in Prisons |
|
|---|---|
| RECOMMENDED |
RATING |
| Prisons should implement opt-out HCV testing. Chronically infected individuals should receive antiviral therapy according to AASLD/IDSA guidance while incarcerated. Upon release, patients should be provided linkage to community healthcare for surveillance for HCV-related complications. | IIa, C |
| To prevent HCV reinfection and reduce the risk of progression of HCV-associated liver disease, prisons should provide harm reduction and evidence-based treatment for underlying substance use disorders. | IIa, C |
Recommendation for Continuation of HCV Treatment in Jail and Prison Settings |
|
|---|---|
| RECOMMENDED |
RATING |
| Jails and prisons should facilitate continuation of HCV therapy for individuals on treatment at the time of incarceration. | IIa, C |
Interventions to reduce HCV transmission and HCV-related liver disease can only be implemented if infected patients are diagnosed. Given the variable approaches to HCV testing across correctional facilities (Maurer, 2015), patients with chronic HCV in these settings may not have the opportunity to be diagnosed (Varan, 2014). Universal opt-out testing of inmates for chronic HCV is highly cost-effective and has been shown to reduce ongoing HCV transmission and the incidence of advanced liver disease (He, 2016). Based on a microsimulation model of HCV transmission and disease progression, this approach would enable diagnosis of 122,700 new HCV infections in prisons in the next 30 years; prevent 12,700 new HCV infections caused by release of infected inmates; and avert 11,700 liver-related deaths (He, 2016).
In October 2016, the Federal Bureau of Prisons recommended an opt-out strategy of testing for HCV infection for all sentenced inmates (FBP, 2016). With this approach, an inmate is informed of the indications and plan for HCV testing, and the test is ordered and performed unless the inmate declines it. However, the Federal Bureau of Prisons clinical guidelines state that HCV testing is not required by policy or law. Thus, it is unclear if prisons are conforming to these recommendations.
HCV-infected individuals in jails frequently cycle in and out of this setting, are unaware of their infection, and can contribute to HCV transmission in the community (Rich, 2014). Therefore, providing opt-out HCV testing in jails followed by linkage to community healthcare providers for those found to be infected is an advantageous approach to HCV case finding in these settings. A recent prospective cohort study evaluated an HCV testing and linkage-to-care program implemented in selected jails in North Carolina and South Carolina from December 2012 to March 2014 (Schoenbachler, 2016). HCV testing and linkage-to-care services were conducted by noncorrectional staff in parallel with correctional healthcare staff. Forty-eight percent of detainees with chronic HCV who were referred for management after release attended a follow-up appointment. Similar programs have been established in New York (Akiyama, 2016), Texas (de la Flor, 2017), and Rhode Island (Beckwith, 2016) with the latter using rapid, point-of-care HCV-antibody testing. These studies demonstrate the feasibility of HCV testing in jails followed by linkage to medical care after release for those who are chronically infected.
A recent observational cohort study demonstrated the feasibility of initiating and completing direct-acting antiviral (DAA) HCV treatment in a jail setting (MacDonald, 2017). In this study, 104 detainees in the New York City jail system received DAA treatment between January 1, 2014 and June 30, 2016, of whom 60% (n=62) entered the jail on DAA therapy and 40% (n=42) initiated DAA treatment in jail. HCV viral loads were undetectable in 94% of community-initiated patients and 97% of jail-initiated patients. This study provides evidence that jail-based initiation of HCV treatment is feasible and prompt access to DAAs in jail can preserve the effectiveness of community-initiated HCV regimens.
HCV DAA therapy for chronic HCV is now logistically feasible within the prison setting and would aid the HCV elimination effort (Spaulding, 2013). The availability of all-oral DAA regimens that commonly require no more than 12 weeks of therapy and cause few adverse effects overcomes many of the logistical challenges associated with interferon-based HCV treatment (Spaulding, 2013). Directly observed therapy is the norm in prison settings, and the risk of drug diversion is low. Returning inmates to their communities cured of chronic HCV would be an invaluable step toward HCV elimination. In addition to these clinical benefits, treating chronic HCV in incarcerated persons is cost-effective. A recent analysis found that sofosbuvir-based treatment for genotype 1 monoinfection met the benchmark for cost-effectiveness in terms of the benefits gained (Liu, 2014).
Given that injection drug use is the major risk factor for initial HCV infection and reinfection, and because alcohol abuse/dependence is a major cofactor in HCV-related liver disease progression, treatment of concomitant substance use disorders along with HCV therapy is of major importance in the incarcerated population. The most effective way to prevent HCV transmission in people who inject drugs is to combine harm reduction strategies that improve the safety of injection (ie, needle/syringe exchange) with interventions that treat the underlying addiction, particularly medication-assisted treatment (MacNeil, 2011); (Volkow, 2014) (see Identification and Management of HCV in People Who Inject Drugs). Alcohol prevention and treatment programs have not been given the same priority as those for drug addiction in correctional settings, and access to treatment for alcohol abuse/dependence after release is often limited. Addressing hazardous alcohol use among inmates with chronic HCV could help slow liver disease progression, decrease HCV transmission, and might reduce recidivism. However, according to the 2015 survey by the American Corrections Association (Maurer, 2015), slightly more than half of correctional systems treat the fundamental substance use disorders among patients receiving HCV antiviral therapy.
To expand HCV testing and prevention counseling and increase access to HCV therapy in correctional institutions, it will be necessary to overcome several important barriers. First, appropriately trained staff are needed to screen inmates for HCV infection and, depending on the result, provide counseling on HCV prevention, linkage to care, and access to antiviral treatment. Offsite providers can assist in these endeavors but add expense and logistical complications. The use of telemedicine to link inmates to specialists has been shown to be effective for the evaluation and treatment of chronic HCV in underserved settings (Arora, 2011). The National Commission on Correctional Health Care supports telemedicine in corrections. However, only 30 of the 45 states responding to the 2016 National Survey of Prison Health Care reported using telemedicine (Maruschak, 2016).
Second, unplanned transfers and releases could disrupt ongoing HCV treatment (Spaulding, 2013). Most state correctional facilities do not have a process in place to smoothly transition a patient receiving DAA treatment in a prison setting to continuing community-based care without a lapse in antiviral therapy. However, the New York State Hepatitis C Continuity Program demonstrated that it is possible to establish a network of community-based providers to facilitate continuation of HCV treatment without interruption after release (Klein, 2007). In this program, inmates who initiated HCV treatment in prison were transitioned to a community-based provider for completion of therapy after release. Inmates diagnosed with chronic HCV who remained untreated while incarcerated were referred to a community provider for treatment evaluation after release.
Finally, the costs of HCV testing and antiviral treatment in correctional facilities are also formidable barriers. Strategies for financing HCV treatment have been put forward by the National Academy of Medicine’s Committee for a National Strategy for the Elimination of Hepatitis B and C (NAS, 2017). These strategies might help overcome cost barriers to HCV testing and treatment in correctional settings.
Addressing these barriers will help ensure that persons residing in jails and prisons can undergo HCV testing and be diagnosed; have access to HCV prevention counseling; and receive treatment for chronic HCV and underlying substance use disorders. Improving the diagnosis and management of HCV infection in correctional settings will greatly facilitate efforts to eliminate HCV infection in the US.